


Der off-label Einsatz könnte die Plörre zum Gentherapieprodukt machen!
The European Regulatory Environment of RNA-Based Vaccines

Im Jahre 2017 schrieben die Gründer von BioNTech (Ugur Sahin und seine Frau Özlem Türeci) zusammen mit dem PEI (Erstautor) ein Buchkapitel darüber, wie man mRNA-Geninjektionen über eine Notfallzulassung auf den Markt bringen könnte.
Hinz T, Kallen K, Britten CM, Flamion B, Granzer U, Hoos A, Huber C, Khleif S, Kreiter S, Rammensee HG, Sahin U, Singh-Jasuja H, Türeci Ö, Kalinke U. The European Regulatory Environment of RNA-Based Vaccines. Methods Mol Biol. 2017;1499:203-222. doi: 10.1007/978-1-4939-6481-9_13. PMID: 27987152. https://pubmed.ncbi.nlm.nih.gov/27987152/

Zu Thomas Hinz ist nicht viel zu finden, nicht einmal auf der PEI Seite. Er scheint aber zum Team Medizinregulatorik zu gehören:

Abstract

“In der EU gibt es derzeit keine regulatorischen Leitlinien, die speziell auf mRNA-basierte Impfstoffe eingehen. Der bestehende Rechtsrahmen legt jedoch eindeutig fest, dass mRNA-basierte Impfstoffe in den meisten Fällen zentral zugelassen werden müssen. Interessanterweise werden RNA-basierte Impfstoffe, je nachdem, ob sie gegen Tumore oder Infektionskrankheiten gerichtet sind, formell als Gentherapieprodukte betrachtet oder nicht. “
Diese regulatorischen Leitlinien gibt es 2025 immer noch nicht.

Das wichtig Stichwort im Abstract lautet übrigens FORMELL!!!!
FORMELL sind mRNA Impfungen keine Gentherapie. Biologisch aber durchaus.
1.1 Die Geschichte der RNA
Das PEI geht, wenn man die hier behandelte Publikation zugrunde legt, wohl von folgenden Voraussetzungen aus:

“Unmittelbar nachdem sie das Zytoplasma erreicht hat, wird sie in Proteine übersetzt, unabhängig davon, ob die mRNA aus dem Zellkern exportiert oder durch eine Transfektionsmethode direkt in die Zelle gebracht wurde. Anders als DNA wird mRNA, die in das Zelllumen gelangt, nicht in das Genom integriert. Daher besteht bei der Transfektion von mRNA nicht einmal ein theoretisches Risiko der Insertionsmutagenese, wie dies bei bestimmten DNA-basierten Gentransferverfahren der Fall ist. Außerdem ist die mRNA, sobald sie das Zytoplasma erreicht hat, nur noch vorübergehend aktiv, da es im Zytoplasma eine Vielzahl physiologischer Mechanismen gibt, die die RNA ständig abbauen.”
Man ging 2017 davon aus, dass bio-mRNA, das was BioNTech uRNA (Uracil-RNA) zum Einsatz kommt. Wenn man davon ausgeht, dass 100,00% uRNA verwendet würde, würde diese Aussage fast stimmen, auch was den schnellen Abbau in der Zelle angeht. Das Problem ist, dass N1-Mythlpseudouridin statt Uracil in der modRNA verbaut wurde und das scheint diese modRNA sehr, sehr, sehr stabil zu machen.1
Fast, weil das Spike-Protein im Endoplasmatischen Reticulum produziert wird2 3 und nicht im Zellumen.

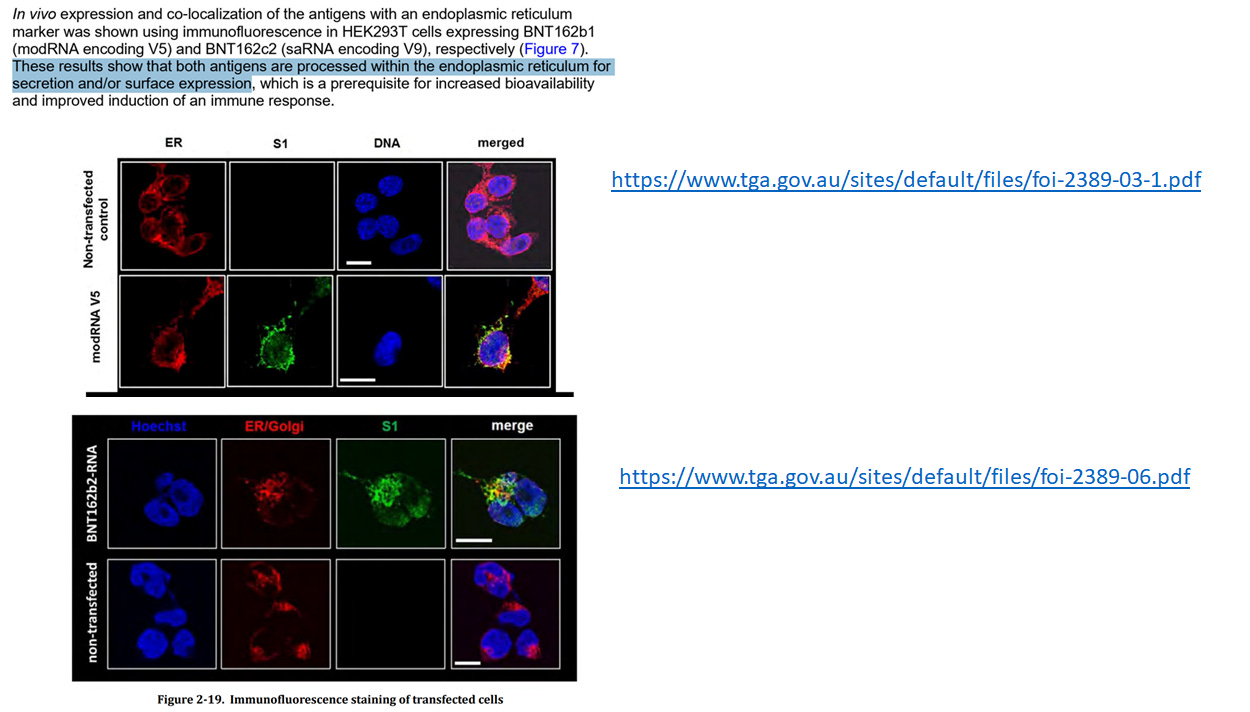
Man gibt aber in dieser Publikation zu, dass DNA insertieren würde und zu Insertionsmutagenese führen würde.
Die COVID-modRNA-Geninjektionen sind jedoch massiv mit DNA-Produktionsrückständen belastet.4
Das Problem ist der EU bekannt5 6 und muss somit auch dem PEI bekannt sein.
1.2 Das therapeutische Potential von mRNA

“eine attraktive Alternative zur Ex-vivo-Produktion von Proteinen, die den Organismus quasi zu seiner eigenen Produktionseinheit macht.”
Hätte damit dem Patienten, der gespritzt wurde, weil er Teil der Produktionskette des biologisch aktiven Endproduktes Spike-Protein war, das Sicherheitsdatenblatt ausgehändigt werden müssen?7

“The mechanism operating after injection of synthetic mRNA in vivo is still unclear, but may involve active transport via receptor-mediated endocytosis or micropinocytosis.”
Der Mechanismus ist 2025 immer noch unklar. 8
1.3 Aktuelle Strategien für RNA-basierte Tumorimpfstoffe

Schauen wir einmal, was 8 Jahre später aus diesen so vielversprechenden Studien geworden ist.



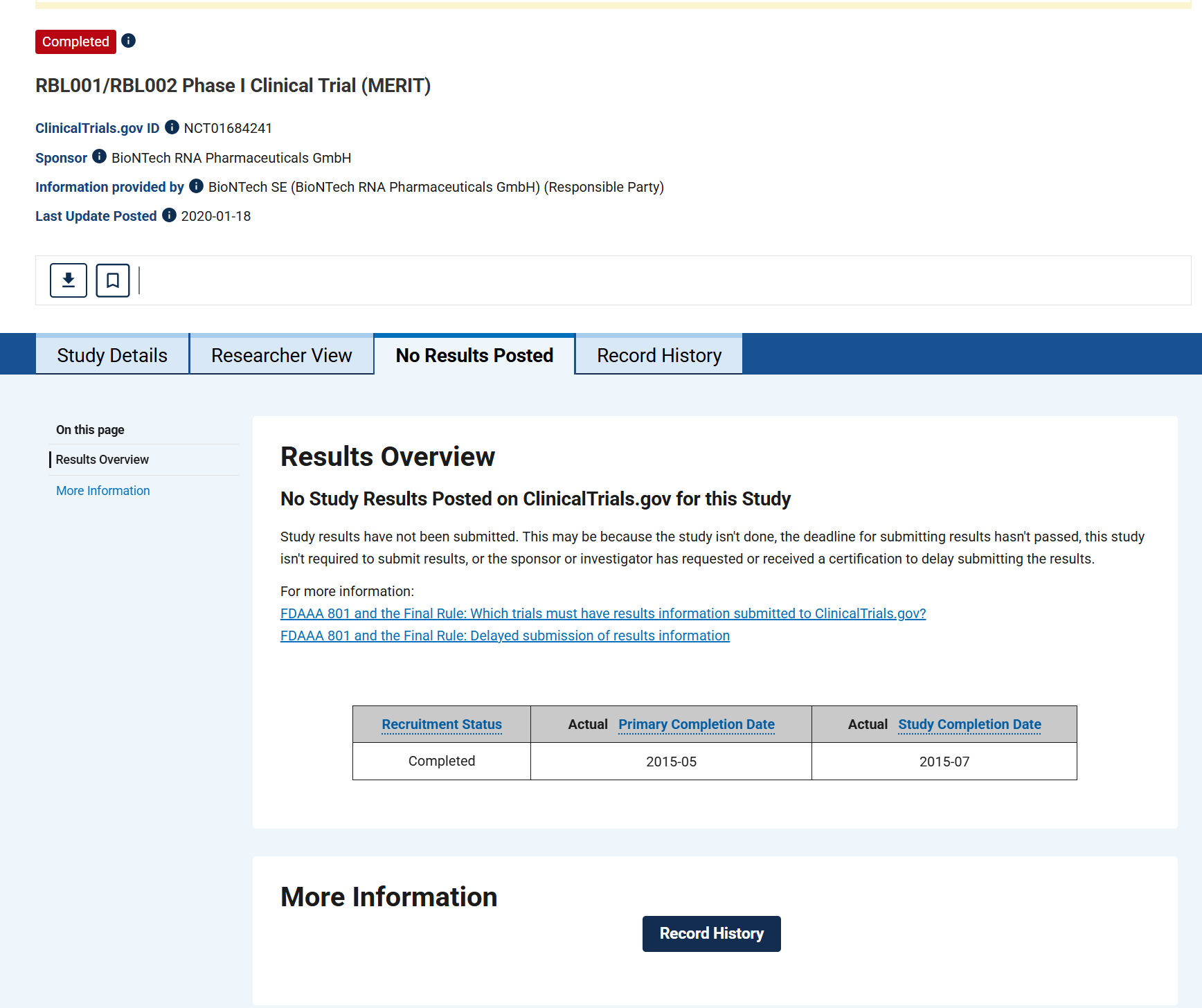
Der Artikel zitiert 3 abgebrochene Studien als potentielle Erfolge, und eine für die bis heute keine Ergebnisse vorliegen. Lief wohl nicht so dolle, sonst hätte man was veröffentlicht?
Mal schauen, ob es hier besser lief:


Gibt es Deadlines, bis wann man seine (negativen) Ergebnisse veröffentlichen muss?

Kleiner Hinweis: BNT162B2 hatte auch dendritische Zellen als Ziel.
„Die Aufgabe dieser neuen Lipide war dieselbe wie jene der Krebsvakzine: die mRNA zu den dendritischen Zellen zu transportieren,“ (Projekt Lightspeed, S. 158)
ABER, weil es keine Krebstherapie war sondern ein Impfstoff, auch wenn es genauso funktionierte, ist BNT162B2 FORMELL keine Gentherapie.
1.4 Aktuelle Strategien für RNA-basierte Impfstoffe gegen Infektionskrankheiten

“Derzeitige Krebsimpfstoffe werden wiederholt und an Orten verabreicht, an denen vermutlich eine hohe Antigenlast vorhanden ist. Impfstoffe gegen Infektionskrankheiten hingegen werden gesunden Menschen prophylaktisch verabreicht, um mit möglichst wenigen Verabreichungen eine schützende Immunreaktion auszulösen. Dies stellt neue Herausforderungen an die Sicherheit und die Induktion einer schützenden Immunreaktion dar. “
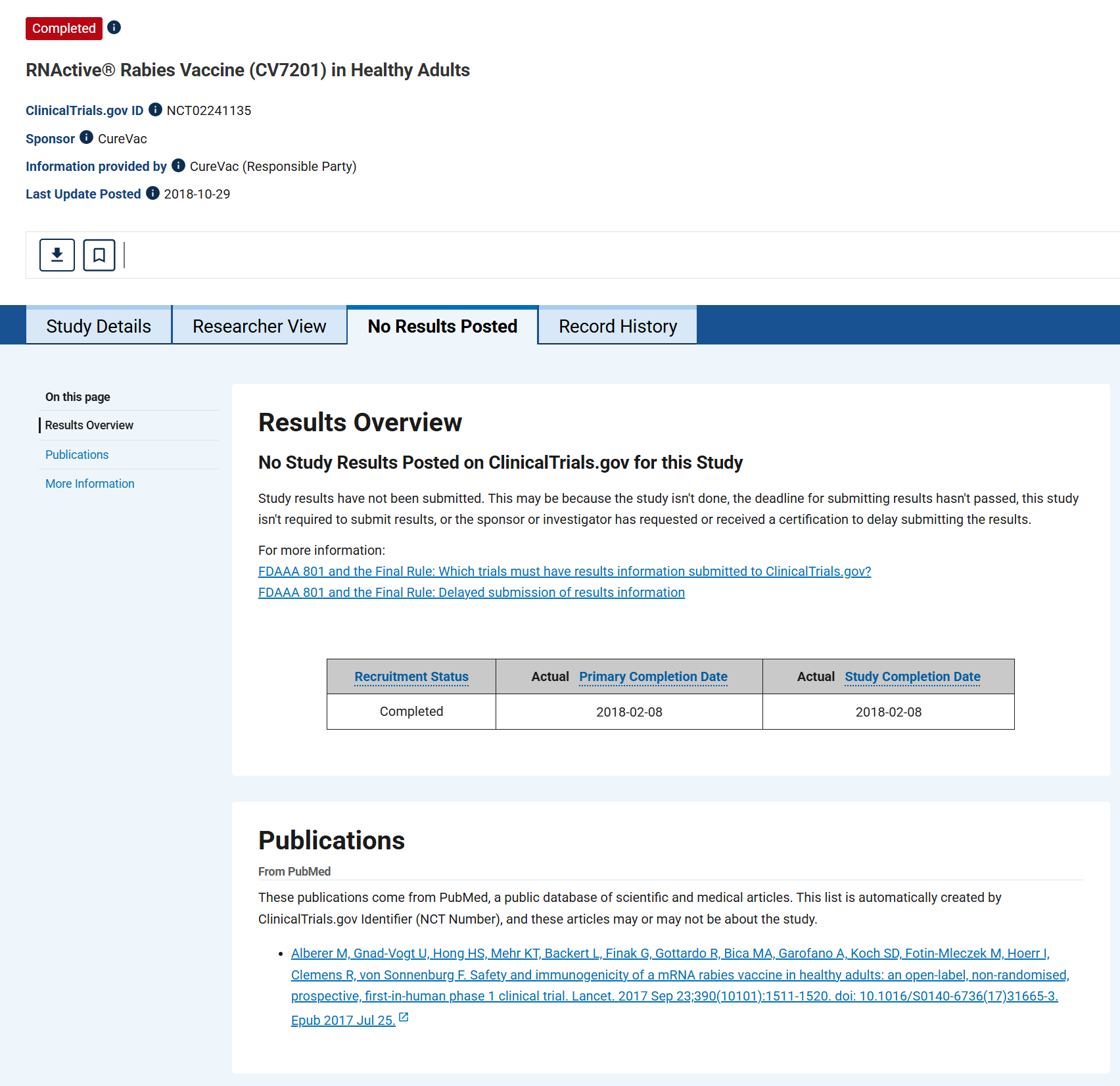
“Diese erstmalige Demonstration am Menschen zeigt, dass ein prophylaktischer mRNA-basierter Impfstoffkandidat verstärkbare funktionelle Antikörper gegen ein virales Antigen induzieren kann, wenn er mit einem nadelfreien Gerät verabreicht wird, jedoch nicht, wenn er mit einer Nadel-Spritze injiziert wird. Der Impfstoff war allgemein sicher und wies ein angemessenes Verträglichkeitsprofil auf.”
Zeitnah publiziert.
Kann es sein, dass, wenn es keine Publikation gibt, der Versuch als gescheitert angesehen werden kann?

Der zitierte HIV Impfstoffversuch war wohl auch ein Rohrkrepierer. Die Webseite existiert nicht mehr.

1.5 Aufkommende therapeutische Optionen für mRNA: Impfstoffe gegen Allergien und In-vivo-Produktion von Proteinen durch mRNA

Dumm nur, dass BNT162B2 anscheinend (laut der Daten verschiedener Impfgeschädigter) einen TH2 Shift ausgelöst hat… Sind die Menschen nun allergisch?
2 The European Regulatory Framework
2.1 EMA und nationale Aufsichtsbehörden

“Neben dem 210 Tage dauernden EMA-Verfahren können die Antragsteller ein beschleunigtes Verfahren beantragen, das nur 150 Tage dauert. Das beschleunigte Verfahren ist anwendbar, wenn das Arzneimittel sowohl aus der Sicht der öffentlichen Gesundheit als auch aus der Sicht der therapeutischen Innovation von großem Interesse ist. Neben dem oben beschriebenen „normalen“ Weg der zentralisierten EMA-Zulassung auf der Grundlage umfassender Daten ist es auch möglich, Produkte auf der Grundlage unvollständiger Daten zuzulassen. Eine solche bedingte Zulassung kann erteilt werden, wenn nur vorläufige klinische Sicherheits- und Wirksamkeitsdaten vorliegen. Auch wenn die klinischen Daten unvollständig sind, muss das von der EMA während des Bewertungsverfahrens ermittelte Nutzen-Risiko-Verhältnis insgesamt positiv sein. Eine weitere Voraussetzung für diesen alternativen Zulassungsweg ist die Fähigkeit des Antragstellers, die fehlenden Daten nach Erteilung der bedingten Zulassung nachzureichen. Außerdem ist das Arzneimittel für Patienten mit ungedecktem medizinischen Bedarf bestimmt, und der Nutzen für die öffentliche Gesundheit muss die Risiken überwiegen, die mit den unvollständigen klinischen Daten verbunden sein könnten. Nach Vervollständigung der Daten kann die bedingte Zulassung in eine reguläre Zulassung umgewandelt werden. Bedingte Zulassungen sind 1 Jahr lang gültig und können verlängert werden.”
Wichtig ist dieser Abschnitt:
Ein weiteres Szenario ist, dass der Antragsteller nicht in der Lage ist, für ein bestimmtes Arzneimittel umfassende Daten zur Unbedenklichkeit und Wirksamkeit unter normalen Anwendungsbedingungen vorzulegen, weil die zu behandelnde Krankheit selten ist oder weil die Erhebung vollständiger Informationen nicht möglich oder unethisch ist. In diesem Fall ist eine Genehmigung für das Inverkehrbringen unter außergewöhnlichen Umständen möglich.“

„Die Aufnahme in die Studie wurde mit unvollständigen Zahlen gestoppt, weil die Rekrutierung langsam war und es unvernünftig/unangemessen wurde, schwangere Frauen nach dem Zufallsprinzip auf Placebo zu setzen, angesichts der Menge an Beobachtungsdaten, die belegen, dass der Impfstoff sicher und wirksam ist, und angesichts der zunehmenden Zahl von Fachausschüssen, die die Immunisierung schwangerer Frauen unterstützen“, schrieb Jelena Vojicic, medizinische Leiterin für Impfstoffe bei Pfizer Canada, in der E-Mail von 2022”
Kommt mir irgendwie bekannt vor. So lief es letztendlich 2020. Und bei den Schwangeren scheint man die Ethikkarte gezogen zu haben.

“Im Gegensatz zu einer bedingten Zulassung ist nicht zu erwarten, dass fehlende Daten nachgeliefert werden können. Daher werden die betroffenen Produkte jährlich überprüft, um ihr Nutzen-Risiko-Verhältnis neu zu bewerten.”

“Wie bereits erwähnt, können die Regulierungsbehörden der einzelnen EU-Mitgliedstaaten als Berichterstatter oder Mitberichterstatter im zentralisierten Verfahren der EMA auftreten.”
Jan Müller-Berghaus vom PEI war er Rapporteur für Modernas Spikevax.10


“Neben ihrer Beteiligung an der EMA sind die nationalen Zulassungsbehörden auch an Tätigkeiten beteiligt, die in die alleinige Zuständigkeit der EU-Mitgliedstaaten fallen. Ein wichtiges Beispiel ist die Genehmigung von Anträgen auf klinische Studien.”

“Die Sponsoren haben die Möglichkeit, einen meldenden Mitgliedstaat vorzuschlagen, der die Hauptprüfung durchführt.”
2.2 Obligatorisches versus optionales zentralisiertes EMA-Zulassungsverfahren

“Zusammenfassend lässt sich sagen, dass für RNA-basierte Arzneimittel, die Gentherapeutika sind (Definition siehe unten), das zentralisierte EMA-Verfahren obligatorisch ist. Sollte jedoch mRNA zum Zweck der Impfung gegen Infektionskrankheiten verwendet werden, handelt es sich per Definition im Gesetz nicht mehr um ein ATMP. Sollte jedoch die rekombinante DNA-Technologie eingesetzt werden, müssen auch solche mRNA-basierten Impfstoffe gegen Infektionskrankheiten über das zentralisierte Verfahren zugelassen werden. Sollten die Kriterien für das zentralisierte Verfahren nicht erfüllt sein, könnten mRNA-Arzneimittel für Infektionskrankheiten über den nationalen Weg zugelassen werden, obwohl der Zugang zum zentralisierten Verfahren auf Antrag des Antragstellers weiterhin möglich ist.”
2.3 Rolle des Ausschusses für neuartige Therapien der EMA bei der Zulassung von mRNA-basierten Arzneimitteln

Gentherapeutikum: ein biologisches Arzneimittel, das folgende Merkmale aufweist:
a) es enthält einen Wirkstoff, der eine rekombinante Nukleinsäure enthält oder aus einer solchen besteht und im oder am menschlichen Körper verwendet wird, um eine genetische Sequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu löschen;
b) seine therapeutische, prophylaktische oder diagnostische Wirkung bezieht sich unmittelbar auf die rekombinante Nukleinsäuresequenz, die es enthält, oder auf das Produkt der genetischen Expression dieser Sequenz.
Zu den Gentherapeutika gehören keine Impfstoffe gegen Infektionskrankheiten.
Also, obwohl ALLE MERKMALE für Gentherapeutika auf die modRNA-Geninjektionen zutreffen, sind die FORMAL keine Gentherapeutika sondern nur biologisch.

“Sollte ein RNA-Molekül auf rein chemischem Wege hergestellt werden, wie es bei vielen RNAi-Molekülen der Fall ist, wäre es kein biologisches Produkt mehr und könnte daher nicht als Gentherapieprodukt eingestuft werden.”
Aus biologischer Sicht kompletter Schwachsinn, regulatorisch mag es Sinn ergeben.

“Wichtig ist auch der Hinweis, dass eine mRNA zur Behandlung oder Vorbeugung von Infektionskrankheiten per Gesetz kein Gentherapieprodukt ist, auch wenn alle anderen Anforderungen erfüllt sind (rekombinant, biologisch). Folglich ist ein mRNA-Molekül, das zur prophylaktischen Impfung, z. B. gegen Influenza, verwendet wird, kein Gentherapieprodukt, während dies beispielsweise bei der Behandlung von Krebs der Fall ist. “
Wer genau hat DAS durchgesetzt und beschlossen und wann?
Die Folgen dieser Gesetze sind dann derart:

Im Vergleich zu Impfstoffen und Immuntherapien erfordern LNP-mRNA-Anwendungen, die keine Immuntherapien sind, eine größere Anzahl präklinischer Tests, wobei der Schwerpunkt auf der Sicherheit liegt. Auf der Grundlage der zuvor erörterten Mechanismen der Immunaktivierung durch LNP-mRNAs würden die präklinischen Tests solcher Studien optimalerweise Folgendes umfassen:
(1) Korrelation einer steigenden LNP-mRNA-Dosis mit der Sekretion von Zytokinen und Chemokinen,
(2) Komplementaktivierung,
(3) Auswirkungen einer wiederholten Verabreichung mit Untersuchung von Anti-Drogen-Antikörpern,
(4) Marker für akute Lebertoxizität und potenzielle Lipidakkumulation und (5) Histopathologie des Zielorgans.
Leider konzentriert sich die derzeitige Literatur über präklinische Studien zur Nicht-Immuntherapie mit LNP-mRNA hauptsächlich auf die Wirksamkeit des Medikaments in Mausmodellen und bietet nur begrenzte Daten zur Sicherheit."
Wie praktisch, dass man sich das nun alles aus JURISTISCHEN Gründen sparen kann, obwohl man weiß, dass es BIOLOGISCH geboten wäre.
Wer hat das geschrieben?
BioNTech selbst:



“Wird mRNA z. B. in DCs transfiziert und nicht direkt an Patienten verabreicht, werden die daraus resultierenden genetisch veränderten Zellen in der Regel ebenfalls als Gentherapieprodukte eingestuft.“
Es sei denn, diese Transfektion findet direkt im Patienten statt, dann ist es keine Gentherapie.

“Wie oben dargelegt, gehören Impfstoffe gegen Infektionskrankheiten nicht zu den Gentherapie-Arzneimitteln. In dem CAT-Reflexionspapier wird jedoch dargelegt, dass ein gentherapeutischer Impfstoff dennoch als Gentherapie eingestuft werden kann, wenn er zur Behandlung oder Vorbeugung von durch die Infektion ausgelösten Krankheiten (z. B. bösartigen Erkrankungen) indiziert ist.“
Heißt das, dass wenn BNT162B2 off-label zur Verhinderung eines schweren Verlaufes eingesetzt wurde, das Produkt doch Gentherapie ist, weil es zur Verhinderung einer Pathologie eingesetzt wurde und nicht zur Prävention?!

“So ist beispielsweise ein mRNA-basierter Impfstoff zur Behandlung oder Vorbeugung von HPV16-induzierten bösartigen Erkrankungen ein Gentherapieprodukt (wenn die Kriterien für Gentherapie erfüllt sind). Wird die identische mRNA für eine Impfung gegen HPV16 verwendet, so wird sie als Impfstoff eingestuft.“

“Die Einzelheiten der mRNA-Herstellung, wie die Verwendung von chemisch synthetisierten Vorlagen im Gegensatz zu aus Bakterien isolierten Plasmid Vorlagen, könnten für die Entscheidung, ob eine mRNA ein biologisches Arzneimittel ist oder nicht, von Bedeutung sein.”

3 Auswirkungen auf mRNA-Impfstoffe
3.2 Regulatorische Qualitätsanforderungen für mRNA

“Die EMA hat bisher noch keine spezifischen Leitlinien für die Entwicklung von mRNA-basierten Arzneimitteln veröffentlicht. Daher müssen die allgemeinen Grundsätze befolgt werden, die in übergreifenden Leitfäden dargelegt sind. Obwohl es sich bei mRNA-basierten Impfstoffen zur Vorbeugung oder Behandlung von Infektionskrankheiten nicht um Gentherapieprodukte handelt, sollten die in der EMA-Leitlinie für Gentherapie-Arzneimittel dargelegten Grundsätze beachtet werden, die sich auf Qualität, nicht klinische und klinische Aspekte beziehen.”

Wie bereits erwähnt, die gibt es 2025 noch nicht.

“Wie bei allen Arzneimitteln muss ein geeignetes Herstellungsverfahren festgelegt werden, das ein Arzneimittel von gleichbleibender Qualität ergibt. Zu diesem Zweck müssen Spezifikationen für kritische Prozessschritte, Zwischenprodukte, Teilprodukte und das Endprodukt festgelegt werden. Auch die Qualität der Roh- und Ausgangsstoffe muss festgelegt und kontrolliert werden.“
Das haben dann letztendlich die Spahn-Gesetze ausgehebelt.14

“Im Allgemeinen sind die zu kontrollierenden Qualitätsmerkmale Aussehen, Identität (und im Falle von Nukleinsäuren die Unversehrtheit), Gehalt, Wirksamkeit, produkt- und verfahrensbedingte Verunreinigungen, Sterilität, Endotoxin und physikalisch-chemische Tests wie pH-Wert und Osmolalität. Sollen mRNA-Moleküle z. B. mit polykationischen Molekülen oder Lipiden komplexiert werden, sollten Tests und Spezifikationen für die Partikelgrößenverteilung festgelegt werden. Die konstante Menge der Komplexbildner sollte entweder Teil der Wirkstofffreisetzung sein oder muss in Validierungsstudien ermittelt werden. […] Neben den Freigabetests sollten während der Produktentwicklung zusätzliche Charakterisierungsstudien mit komplexierten Nukleinsäuren durchgeführt werden, die sich mit Eigenschaften wie Form, Oberflächenladung und Stabilität befassen.”
Die Lipide bilden Addukte15 und die Oberflächenladung wurde nicht gemessen.

GMP… Da war doch was:


“Die letztgenannte Referenz enthält jedoch keine Definition des Zeitpunkts, ab dem GMP für Plasmide und nichtvirale Vektoren erforderlich ist. […] Obwohl es von Vorteil sein könnte, mikrobielle Zellbänke in das GMP-System einzubeziehen, gibt es derzeit keine klaren Anforderungen. Andererseits stellen die verfügbaren Leitlinien keine GMP-Anforderungen für die rekombinante Technologie, die für die anfängliche Plasmid-/Template-Herstellung verwendet wird. Es besteht jedoch kein Zweifel daran, dass es sich bei der In-vitro-Transkription um die Herstellung von Arzneimitteln handelt, die unter GMP durchgeführt werden muss.“
Diese Zweifel wurden dann wohl zu Gunsten der Hersteller ausgelegt und man hat GMP ignoriert?
3.3 Präklinische regulatorische Anforderungen für mRNA-Arzneimittel

“Impfstoffe werden in der Regel lokal über eine intradermale oder intramuskuläre Injektion verabreicht. Pharmakokinetische Studien sind daher normalerweise nicht erforderlich [63]. Andererseits werden mRNAs, die für die therapeutische Krebsimpfung bestimmt sind, manchmal systemisch über den intravenösen Weg verabreicht. Bei einmaliger oder wiederholter systemischer Verabreichung scheint die Bewertung der pharmakokinetischen Parameter sowohl unter Sicherheits- als auch unter Wirksamkeitsaspekten wichtig zu sein. […]
Dies wiederum könnte bedenklich sein, da mRNA per se immunstimulierend ist und die Sekretion von entzündungsfördernden Zytokinen auslösen kann. Aufgrund der Fähigkeit mehrerer Gentherapeutika, sich in das Genom des Empfängers zu integrieren, werden Studien zur Biodistribution normalerweise als wichtig erachtet [59]. […]
Dies könnte anders sein, wenn chemisch veränderte Nukleoside verwendet werden.”
Windelweich herausgewunden, ob man bei modRNA nun Pharmakokinetik machen muss oder nicht.
3.4 Klinische Zulassungsanforderungen für mRNA-Medikamente

“Interessanterweise werden in der Leitlinie (für mRNA Impfstoffe) keine pharmakokinetischen Studien gefordert, obwohl Nukleinsäuren eingeschlossen sind. Im Gegenteil, pharmakokinetische Studien werden für Gentherapeutika erwartet.“
Man kann es durchaus als “interessant” bezeichnen, dass mit zweierlei Maß gemessen wird. Mein bevorzugter Begriff wäre “idiotisch” oder “potentiell tödlich”.
Meine Schlussfolgerung
Juristisch könnte dieses PEI Paper vielleicht Hinweise geben, wie man aufgrund der Herstellung nach Verfahren 2 und des von der Politik propagierten off-Label use zur Verhinderung schwerer Verläufe, die COVID-Geninjektionen auch juristisch wieder zu Gentherapien machen könnte.
Das ist aber Aufgabe der Juristen. An der Stelle bin ich raus.
Ich habe nur die Highlights zusammengengefasst, weil die mir bekannten Juristen seit Jahren keine Zeit/Lust hatten, dieses Buchkapitel durchzuarbeiten.
Vitriol, D. V. (2025ah, April 4). modRNA noch ca. 17 Monate später in Hirnproben nachweisbar. DrBine’s Newsletter.
modRNA noch ca. 17 Monate später in Hirnproben nachweisbar

Die Japaner haben mal wieder eine Studie veröffentlicht, die weiteres Licht auf die Schadmechanismen der modRNA-Plörren wirft.
Vitriol, D. V. (2025k, March 17). Slowakische Daten zu DNA-Produktionsrückständen. DrBine’s Newsletter.
Slowakische Daten zu DNA-Produktionsrückständen

Langsam verliert man den Überblick, wer schon alles die Verunreinigungen durch DNA Rückstände aus der Produktion bestätigt hat.
Vitriol, D. V. (2023v, December 26). Die EMA weiß, dass der DNase I - Verdau nicht funktioniert und hier sind die Dokumente, die das belegen. DrBine’s Newsletter.
Die EMA weiß, dass der DNase I - Verdau nicht funktioniert und hier sind die Dokumente, die das belegen

Diese Daten sind so brisant, dass ich sie einzeln auslager, weil sie in meinen Artikeln als Teilaspekt unterzugehen scheinen.
Vitriol, D. V. (2024ak, December 3). Pfizer gibt den SV40 Promotor in EMA Dokumenten zu. DrBine’s Newsletter.
Pfizer gibt den SV40 Promotor in EMA Dokumenten zu

Tobias Ulbrich hat am 1. Dezember folgendes getwittert:
Vitriol, D. V. (2025r, March 23). Das Comirnaty Sicherheitsdatenblatt. DrBine’s Newsletter.

Vitriol, D. V. (2025am, April 7). Wie entkommen die LNPs aus den Endosomen? (Endosomal Escape). DrBine’s Newsletter.

Stieber, Z. (2023, March 1). Pfizer confirms it ended COVID-19 vaccine pregnancy trial early. The Epoch Times. https://www.theepochtimes.com/health/pfizer-says-it-ended-covid-19-vaccine-pregnancy-trial-early-5088375
Vitriol, D. V. (2024a, January 16). Wie man sich aus seinen eigenen Worten gemeinsam einen Strick dreht wissen das PEI und der Klausi Teil 1. DrBine’s Newsletter.
Wie man sich aus seinen eigenen Worten gemeinsam einen Strick dreht wissen das PEI und der Klausi Teil 1

Im Mai 2021 reichte die Arbeitsgruppe von/um Klaus Cichutek einen wissenschaftlichen Artikel über die Covid-Injektionen beim Journal Vaccines ein, welches im Juni 2021 veröffentlicht wurde.
Vlatkovic I. Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines. 2021 May 10;9(5):530. doi: 10.3390/biomedicines9050530. PMID: 34068715; PMCID: PMC8151051. https://pubmed.ncbi.nlm.nih.gov/34068715/
Vitriol, D. V. (2023q, November 6). Die kurios verquere Gedankenwelt der modRNA Gläubigen Teil 2. DrBine’s Newsletter.
Die kurios verquere Gedankenwelt der modRNA Gläubigen Teil 2

Hier ein weiterer Artikel aus der Reihe: Die kurios verquere Gedankenwelt der modRNA Gläubigen.
Vitriol, D. V. (2024ao, December 9). Die Bundesregierung setzt heimlich Gesetze und Verordnungen für Covid-19-Impfstoffe außer Kraft. DrBine’s Newsletter.
Die Bundesregierung setzt heimlich Gesetze und Verordnungen für Covid-19-Impfstoffe außer Kraft

Edgar hat das sehr schön zusammengestellt, hat aber keinen eigenen Substack.
Packer M, Gyawali D, Yerabolu R, Schariter J, White P. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat Commun. 2021 Nov 22;12(1):6777. doi: 10.1038/s41467-021-26926-0. PMID: 34811367; PMCID: PMC8608879. https://pubmed.ncbi.nlm.nih.gov/34811367/
Vitriol, D. V. (2023x, December 26). Die EMA weiß, dass der DNase I - Verdau nicht funktioniert und hier sind die Dokumente, die das belegen. DrBine’s Newsletter.
Die EMA weiß, dass der DNase I - Verdau nicht funktioniert und hier sind die Dokumente, die das belegen
Diese Daten sind so brisant, dass ich sie einzeln auslager, weil sie in meinen Artikeln als Teilaspekt unterzugehen scheinen.
Ich bin begeistert von Ihrer fachlichen Kompetenz
Also ich finde es äußerst perfide, wie durch Interpretation von Gesetzesauslegungen hergeleitet wird, dass die Spritzplörre auf keinen Fall getestet werden muß. Parallel dazu setzt sich ein Gesundheitsminister z.B. mit Alena B. vor eine Kamera und behauptet einfach mal so, kein anderes Medikament sei jemals so gut untersucht worden. Wie nennt man sowas, Chuzpe?
Was wurde hinter verschlossenen Türen noch alles besprochen, um diesen Coup durchzuziehen? Ich dachte bisher, die Meldung, dass eine Biowaffe freigesetzt worden sei oder durch einen Unfall freigesetzt wurde, war der Treiber. Aber letztendlich passt das doch nicht in den zeitlichen Ablauf. Die Änderungen des Arzneimittelgesetzes, zur Ausklammerung von modRNA Medikamenten, geschah ja schon 2009. Spahn hat ja nur noch ein paar Falten im Gesetz glattgebügelt und Steine aus dem Weg geräumt, die die ganze Sause noch hätten gefährden können oder?
Meiner Meinung nach und das ist aber auch nur Spekulation, war die Triebfeder in Deutschland BioNTech, also Ugur Sahin und seine Frau, während Spahn und Merkel angeschissen wurden mit dem Märchen von der tödlichen Biowaffe, weshalb alles und jeder der nicht „eingeweiht“ war und nur den gesunden Verstand hatte, zum Schweigen gebracht werden musste. Das ist doch alles bestimmt nur wilde Verschwörungstheorie, so menschenverachtend kann doch niemand sein oder? Und doch fallen mir Namen ein. Meiner Frau sage ich immer Grünenthal und Contergan waren nur laue Lüftchen gegen das, was da gelaufen ist. Andere Beispiele mag ich hier nicht nennen.
Ich bin jedoch zu 100% sicher, dass es nicht passieren darf, dass durch schlampige Produktion fahrlässig Menschen gefährdet werden dürfen. Als ich noch in der chemischen Industrie gearbeitet habe, war ich gemeinsam mit unserer Produktsicherheitsabteilung und den Produktionsbetrieben erpicht darauf alles was ich so entwickelt hatte und was mit Menschen in Kontakt kommen sollte, bis ins Detail untersucht wurde.
Und ich war nur für die Entwicklung und Produktion von speziellen Textilfarben verantwortlich. Pharma war m.E. eine ganz andere Stufe.