


Wie man sich aus seinen eigenen Worten gemeinsam einen Strick dreht wissen das PEI und der Klausi Teil 2
Klaus Cichutek (2021) - Accelerated Development of COVID-19 Vaccines

Nachdem wir in Teil 1 den Bias der Autoren durchleuchtet haben,
Wie man sich aus seinen eigenen Worten gemeinsam einen Strick dreht wissen das PEI und der Klausi Teil 1

Im Mai 2021 reichte die Arbeitsgruppe von/um Klaus Cichutek einen wissenschaftlichen Artikel über die Covid-Injektionen beim Journal Vaccines ein, welches im Juni 2021 veröffentlicht wurde. Wagner R, Hildt E, Grabski E, Sun Y, Meyer H, Lommel A, Keller-Stanislawski B, Müller-Berghaus J, Cichutek K. Accelerated Development of COVID-19 Vaccines: Technology…
widmen wir uns in Teil 2 dem eigentlichen Paper.
Was behauptet das PEI in dieser Publikation, wie die Zulassung der Covid-Injektionen angeblich verlaufen ist und was sagen die offiziellen, herausgeklagten Unterlagen, wie es wirklich gelaufen ist?
Dieses Paper wurde im Juli 2021 veröffentlicht. Alle Autoren arbeiten im und für das PEI. Zu diesem Zeitpunkt ging man beim PEI sicherlich davon aus, dass diese behördliche Darstellung der Ereignisse unangefochten so stehenbleiben würde. Keiner rechnete damit, dass man die originalen Studiendokumente herausklagen würde und dass die EU-Verträge, bzw. die Rolling-Reviews der EU jemals ans Licht der Öffentlichkeit gelangen würden. Man fühlte sich sicher und schrieb sich die Realität herbei, wie sie die wissenschaftliche Gemeinschaft und die Öffentlichkeit glauben sollte.
Ich führe daher in diesem Artikel einen Realitätscheck durch.
Was hat das PEI behauptet und was ist wirklich passiert.
Ich beschränke mich dabei, wie gewohnt, auf die Highlights des Papers.
Wagner R, Hildt E, Grabski E, Sun Y, Meyer H, Lommel A, Keller-Stanislawski B, Müller-Berghaus J, Cichutek K. Accelerated Development of COVID-19 Vaccines: Technology Platforms, Benefits, and Associated Risks. Vaccines (Basel). 2021 Jul 6;9(7):747. doi: 10.3390/vaccines9070747. PMID: 34358163; PMCID: PMC8310218. https://pubmed.ncbi.nlm.nih.gov/34358163/
Wer es genau wissen will, muss das gesamte Werk selbst lesen. Es ist kostenlos und frei herunterladbar.
Zitate des PEI und anderer Publikationen werden dabei, wie gewohnt, kursiv in Englisch und Deutsch angeben, damit sie jeder im Originaltext finden und überprüfen kann.
Abstract
EN: “The acceleration in vaccine development required to combat the current pandemic is not at the expense of the necessary regulatory requirements, including robust and comprehensive data collection along with clinical product safety and efficacy evaluation.
DE: "Die Beschleunigung der Impfstoffentwicklung, die zur Bekämpfung der aktuellen Pandemie erforderlich ist, geht nicht auf Kosten der notwendigen regulatorischen Anforderungen, einschließlich einer soliden und umfassenden Datenerhebung sowie einer klinischen Bewertung der Produktsicherheit und -wirksamkeit.“
Schon an diesem einzelnen Satz des Abstracts kann man sich ordentlich abarbeiten.
Die Zulassung ging also nicht auf Kosten der notwendigen regulatorischen Anforderungen?
Beim verhunzten DNase I – Verdau ist die EMA einfach eingeknickt und hat aufgegeben.
Die EMA weiß, dass der DNase I - Verdau nicht funktioniert und hier sind die Dokumente, die das belegen

Diese Daten sind so brisant, dass ich sie einzeln auslager, weil sie in meinen Artikeln als Teilaspekt unterzugehen scheinen. DIE EMA WEISS, DASS DER DNASE I -VERDAU NIE FUNKTIONIERT HAT UND IMMER NOCH NICHT FUNKTIONIERT UND HAT DAS AKZEPTIERT!!!!!!!!!!!!!!
Ein Großteil der verwendeten Chemikalien ist nicht in der Europäischen Pharmacopeia eingetragen:

Man hat Tote zu spät gemeldet, damit man seine Notfallzulassung bekommt, die man sonst nicht bekommen hätte, weil es im Verum mehr Tote gab, als im Placebo (https://ijvtpr.com/index.php/IJVTPR/article/view/86):
https://drbine.substack.com/p/der-heie-schei-teil-4-todesmeldungen
Die Hütchenspiele bei der Dosierung und bei den Varianten?
Zu all dem komme ich später im Text noch (immer wieder) und detaillierter. Aber streng genommen, kann man das auch bereits alles im Abstract abhandeln, weil bereits dieser all die großen Lügen des PEI in kondensierter Form enthält.
Was die umfassenden Datenerhebung, klinischen Bewertung der Produktsicherheit und -wirksamkeit angeht:
Umfassende Datenerhebung und klinischen Bewertung der Produktsicherheit beim PEI:
1. Die SaveVac-App Daten sind bei heute nicht verfügbar für die Allgemeinheit.
2. Man hat es nicht geschafft, eine Schnittstelle zu den KV-Daten zu programmieren (https://twitter.com/DrDrAlexanderW1/status/1711452567538901194)
3. Man hat die falsche Methode verwendet, um Sicherheitssignale überhaupt bemerken zu können (Observed vs. Expected, totaler Humbug ohne passender Referenzgruppe bei spontanen Berichten. Dreister Betrug)

„Dem Paul-Ehrlich-Institut liegen keine Informationen vor, wie viele Chargen in Deutschland tatsächlich verimpft wurden. Aussagen sind möglich zur Zahl ausgelieferter Chargen, seit die Distribution der COVID-19-Impfstoffe mit Wirkung zum 01.05.2022 von der Bundeswehr auf das Zentrum für Pandemie-Impfstoffe und -Therapeutika (ZEPAI) am Paul-Ehrlich-Institut übergegangen ist. Für den Zeitraum vor dem 01.05.2022 wenden Sie sich bitte an die Bundeswehr.“ (https://fragdenstaat.de/anfrage/todesfaelle-covid-19-impfung/#nachricht-820720)
Wie genau berechnet man die Sicherheit, wenn man nicht einmal weiß, wie viele Chargen in Deutschland verimpft wurden. Da fehlt einem doch irgendwie die Referenzmenge für die Berechnung der Sicherheit?

„Der teilnehmende Mitgliedsstaat erkennt an, dass der Impfstoff und die mit dem Impfstoff zusammenhängenden Materialien sowie ihre Komponenten und Bestandteile aufgrund der Notsituation der COVID-19-Pandemie rasch entwickelt und nach der Bereitstellung des Impfstoffs an die teilnehmenden Mitgliedstaaten im Rahmen des APA weiter untersucht werden. Der teilnehmende Mitgliedsstaat erkennt ferner an, dass die langfristigen Auswirkungen und die Wirksamkeit des Impfstoffs derzeit nicht bekannt sind und dass der Impfstoff unerwünschte Wirkungen haben kann, die derzeit nicht bekannt sind. Weiterhin, soweit anwendbar, erkennt der Teilnehmerstaat an, dass der Impfstoff nicht in Serie produziert werden wird.” https://d7694293-ffb8-4ed0-a014-3581d49070e4.usrfiles.com/ugd/d76942_5af19ff7389d405585ae0c9db50eb306.pdf (S. 17 Artikel 1)
Die nicht stattgefundenen Chargenprüfungen sind noch mal ein Thema für sich.
Zumindest die freigeklagten Studienunterlagen hätte sich das PEI mittlerweile ansehen können, um die Analysen zu bestätigen oder zu widerlegen, die andere durchgeführt haben. Man kam auf Rate von schweren Nebenwirkungen von 10,1 AESIs gegenüber 2,3 Hospitalisierungen wegen COVID bei 10 000 Teilnehmern (https://www.sciencedirect.com/science/article/pii/S0264410X22010283?via%3Dihub).
10,1/2,3= 4,4
4,4x mehr schwere Impfschäden als Hospitalisierungen bei schwerem Verlauf. DAS klingt so richtig sicher und nach positivem Kosten:Nutzen-Verhältnis.
Obwohl rein finanziell ist das Kosten:Nutzen-Verhältnis für den Pharmazeutisch-medizinischen-
Was die Wirksamkeit angeht… wie genau wurde die ohne Placebo-Gruppe bestimmt? Die Wirksamkeitsstudie konnte somit nie abgeschlossen werden und wird somit wohl die Deadline, wegen fehlender Placebogruppe verfehlen. Zumal es den harten Endpunkt der Verhinderung von Todesfällen gar nicht gab. Wenn man diese Daten nicht erhebt, wie will man dann eine Wirksamkeit berechnen? Dass die Übertragung nicht verhindert wird, hat man ja beim RKI bereits im März 22 zugegeben (https://media.frag-den-staat.de/files/foi/778388/23-02-2023-aw-rki-220322-rki.pdf) und dafür war das Produkt letztendlich von der EMA auch nicht zugelassen. Meint man mit Wirksamkeit vielleicht den (nicht gegebenen) Selbstschutz?
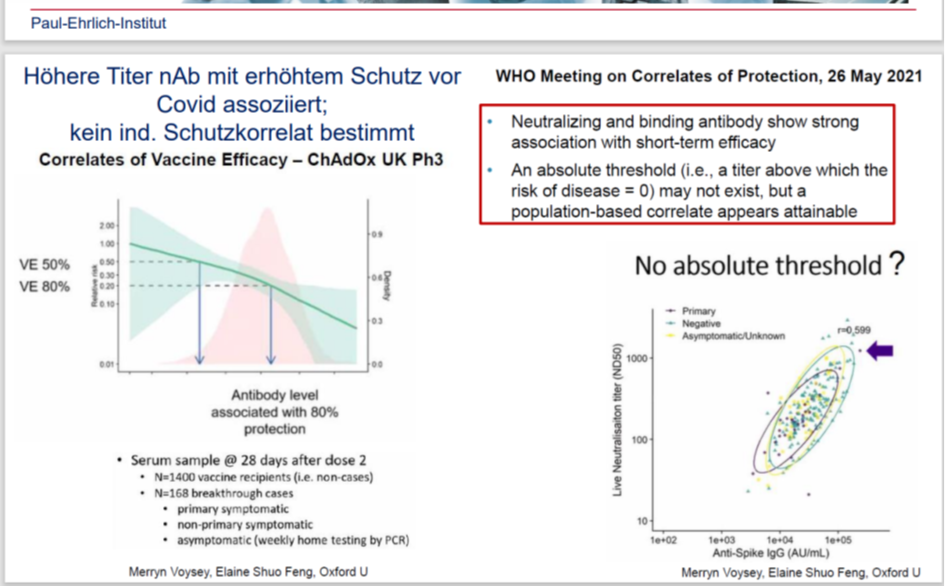

https://twitter.com/Sabisteb/status/1746999459433570553
Professor Norman Fenton erklärt die statistischen Tricks, die für die Schrottwissenschaft unter anderem auch des PEI angewandt wurden.

Ich will an dieser Stelle nicht zu sehr vorgreifen. Wir sind ja noch im Abstract.
EN: “Due to the previous development of vaccine candidates against the related highly pathogenic coronaviruses SARS-CoV and MERS-CoV, the antigen that elicits immune protection is known: the surface spike protein of SARS-CoV-2 or specific domains encoded in that protein, e.g., the receptor binding domain.
DE: „Aufgrund der früheren Entwicklung von Impfstoffkandidaten gegen die verwandten hochpathogenen Coronaviren SARS-CoV und MERS-CoV ist das Antigen, das den Immunschutz auslöst, bekannt: das Oberflächen-Spike-Protein von SARS-CoV-2 oder spezifische Domänen, die in diesem Protein kodiert sind, z. B. die Rezeptorbindungsdomäne.“
NEIN. Schaut man in alte Virologiebücher aus dem Jahr 2016, findet man auf S. 445 der 5. Auflage von Fenner and White’s Medical Virology:

Das ist eine höfliche Art zu schreiben, dass die S-Proteine dieser Viren derartig schnell mutieren, dass sich mit ihnen keine langfristige Immunität erreichen lässt.
Gut, das geht auch einfacher als mit einem Virologiebuch, nämlich mit der Anleitung zum Abwassertest des RKI:

Das Spike mutiert zu schnell für einen sicheren Abwassertest, daher ist es das perfekte Protein für langfristige Immunität, schon klar.
Hat hier das RKI das PEI argumentativ vor dem Bus gestoßen?
Oder sagen wir es doch einfach mit Pfizers eigenen Worten aus einem ihrer Preprints:

Das Bild ist der Beweis, das es hier nicht um in „Schwurblerpaper“ geht, sondern um eines des Herstellers selbst.

„Ältere Versionen der COVID-19-Impfstoffe boten, wenn überhaupt, nur einen geringen zusätzlichen Schutz im Vergleich zu Ungeimpften, auch vor COVID-19-Krankenhauseinweisungen, unabhängig von der Anzahl oder Art der zuvor erhaltenen Dosen“ https://www.medrxiv.org/content/10.1101/2023.12.24.23300512v1
Deren Worte, nicht meine!
Also zurück auf Start für alle, die mitgemacht haben!
Willkommen im jährlichen Abbo, um trotzdem bei der Hospitalisierung schlechter dazustehen, als jene, die nicht mitgemacht haben. Kann aber auch einfach daran liegen, dass Ungeimpfte Krankenhäuser und Ärzte mittlerweile generell meiden.
Wie war das mit 95% Schutz mit nur 1-2 Spritzen. OK, ein Booster. Willkommen im jährlichen Abbo.
Das gibt spannende hybride Trimere, wenn der Rest an Untereinheiten des Proteins noch von einigen Zellen weiter produziert wird.
4-5 Varianten ein Monomeren bei einem Trimeren Protein, das Hexamere und Nonamere bilden (https://doi.org/10.1126/science.abe1502) kann. Kombinatorik Modell 5 Kugeln von denen 3x gezogen wird, mit jeweils zurücklegen? Wäre jetzt meine Vermutung für die zu verwendende Formel. Bei Hexameren und Nonameren wäre das 6 oder 9 mal ziehen mit zurücklegen. Das sind echt viele Möglichkeiten und es werden mit jeder neuen Injektion mehr. Da bekomme ich einen Knoten im Hirn.
Die Cleveland-Klinik Studie hat bereits im April 2023 sehr deutlich gezeigt, dass die Covid-Injektionen die Infektionswahrscheinlichkeit erhöht haben und zwar umso mehr, umso öfter geboostert wurde. Das geringste Risiko hatten die Ungeimpften.

(https://academic.oup.com/ofid/article/10/6/ofad209/7131292).
Netterweise hat man bei diesem Bild auch gleich eingetragen, welche Variante wann dominant war. BioNTech/Pfizer hingen mit ihrem Produkt immer der Natur hinterher, damit war nie wirklich Schutz gegeben und das gibt Pfizer indirekt in seinem Preprint auch zu, so zwischen den Zeilen. Obwohl die Hauptaufgabe der Studie natürlich war, denn (Un-)Sinn des Dauerabos zu belegen.
Zudem wusste man bereits von SARS, dass das Spike-Protein toxisch ist und daher eine echt schlechte Wahl, ganz abgesehen davon, dass es zu schnell mutiert, um überhaupt eine langfristige Immunität zu erzeugen.
2007 wusste man bereits, dass das Spike-Protein stark inflammatorisch wirkt und zur Erhöhung der Entzündungsmarker IL-6 and TNF-alpha führt (https://pubmed.ncbi.nlm.nih.gov/17532082/), an Makrophagen bindet und diese aktiviert (https://pubmed.ncbi.nlm.nih.gov/17412287/), Stress im Endoplasmatischen Retikulum verursacht (wo es in der Zelle auch vom BioNTech/Pfizer Produkt hergestellt wird) und die Chemokin-mRNA Konzentration dadurch zunimmt (https://pubmed.ncbi.nlm.nih.gov/17670839/) und dass das Spike-Protein an ACE2 bindet und damit das Renin-Angiotensin System stört (https://pubmed.ncbi.nlm.nih.gov/17558469/). DAS ist nur eine winzige Auswahl der Probleme, die man über das Spike Protein von SARS bereits vor fast 20 Jahren wusste. Das und der Hinweis, dass das Spike-Protein so schnell mutiert, dass man keine langfristige (natürliche) Immunität dagegen aufbaut, machten es somit zum idealen Kandidaten für die Covid-modRNA-Produkte.
Streng genommen lügt das PEI nicht direkt, wenn es schreibt: „ist das Antigen, das den Immunschutz auslöst, bekannt: das Oberflächen-Spike-Protein von SARS-CoV-2 oder spezifische Domänen, die in diesem Protein kodiert sind, z. B. die Rezeptorbindungsdomäne.“
Man hätte halt nicht „das Antigen“, sondern „ein Antigen“ schreiben müssen, weil das Nucleocapsid das auch macht und nicht so schnell mutiert und nicht so schädlich ist, und man hätte vielleicht in diesem Satz die Toxizität dieses Proteins ansprechen sollen. Unterschlagen/Verschweigen ist eine Form der Lüge.
Es überrascht daher nicht wirklich, dass das in der modRNA-kodierte Spike-Protein, sich genauso toxisch, wenn nicht toxischer verhält, als seine Vorfahren (https://www.mdpi.com/2227-9059/11/8/2287). Dieser Substack soll aber kein Review über die Toxizität des Spike-Proteins werden, dazu gibt es schon genug wissenschaftliche Literatur (z. Bsp. https://pubmed.ncbi.nlm.nih.gov/37392949/, https://pubmed.ncbi.nlm.nih.gov/37317282/).
EN: “From a scientific point of view and in accordance with legal frameworks and regulatory practices, for the approval of a clinic trial, the Paul-Ehrlich-Institut requires preclinical testing of vaccine candidates, including general pharmacology and toxicology as well as immunogenicity.”
DE: „Aus wissenschaftlicher Sicht und in Übereinstimmung mit den gesetzlichen Rahmenbedingungen und der behördlichen Praxis verlangt das Paul-Ehrlich-Institut für die Genehmigung einer klinischen Studie präklinische Tests von Impfstoffkandidaten, einschließlich der allgemeinen Pharmakologie und Toxikologie sowie der Immunogenität".
Das PEI hat also allgemeine Pharmakologie, Toxikologie sowie Immunogenitätsdaten verlangt. Die hätte ich dann sehr gerne. Das PEI wäre nämlich weltweit die einzige Behörde, die diese Daten hat.

Obwohl Immunogenitätsdaten gibt es schon, nur… man weiß halt nicht genau, was sie bedeuten, weil man keine Ahnung hat, wie das Produkt vom immunologischen Mechanismus her funktioniert.

„Der exakte immunologische Mechanismus, der für Schutz gegen SARS-COV-2 verantwortlich ist, ist unbekannt“( https://www.fda.gov/media/151733/download (S. 15))
Wenn man keine Ahnung hat, wie das Produkt immunologisch überhaupt funktioniert? Woher weiß man dann, welche Werte man messen muss und was diese Werte in Relation zum (unbekannten) Wirkmechanismus bedeuten? Ich frag nur für einen Freund.
1. Einleitung
EN: “Worldwide, agreements have been made between the globally active medicines regulatory agencies on the criteria for the approval of clinical trials. Creating the necessary and appropriate balance between possible regulatory simplifications and essential requirements is a scientific task, the basics of which the Paul-Ehrlich-Institut, as the Federal Institute for Vaccines and Biomedical Medicines, has summarized in the following text.”
DE: "Weltweit haben sich die global tätigen Arzneimittelzulassungsbehörden auf die Kriterien für die Genehmigung klinischer Prüfungen geeinigt. Die notwendige und angemessene Balance zwischen möglichen regulatorischen Vereinfachungen und notwendigen Anforderungen zu schaffen, ist eine wissenschaftliche Aufgabe, deren Grundlagen das Paul-Ehrlich-Institut als Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel in dem folgenden Text zusammengefasst hat."
Das Thema „Balance zwischen möglichen regulatorischen Vereinfachungen und notwendigen Anforderungen zu schaffen“ habe ich bereits in diesem Artikel ausführlich behandelt:
Wie man sich aus seinen eigenen Worten einen Strick dreht, weiß auch das PEI Teil 2

Das PEI hat neben Dr. Sediri-Schöns Paper (https://link.springer.com/article/10.1007/s00103-022-03611-1) auch andere interessante Publikationen verfasst, welche beschreiben, worin das PEI seine eigentliche Aufgabe sieht, und die ist nicht der Verbraucherschutz, um es mal vorsichtig zu spoilern.
Da höre ich Pfleiderer und Wichmann in diesen Zeilen. Sie haben es aber ein wenig anders formuliert:
„Auf Grundlage dieser Überlegungen ist in den vergangenen Jahren das Konzept der regulatorischen oder auch zulassungs- und prüfungsbegleitenden Forschung entstanden. Dieses fordert, dass sich sowohl eine für Impfstoffe zuständige nationale Behörde wie das Paul-Ehrlich-Institut (PEI) als auch die Europäische Arzneimittelagentur (European Medicines Agency, EMA) als Mediatoren verstehen, die auf der Basis wissenschaftlich fundierter Untersuchungen die Balance zwischen gesetzgeberischen und regulatorischen Notwendigkeiten und medizinischen Problemstellungen finden. (https://www.pei.de/SharedDocs/Downloads/wiss-publikationen-volltext/bundesgesundheitsblatt/2015/2015-zulassung-impfstoffe-empfehlung-stiko-kriterien-nutzen-risiko.pdf?__blob=publicationFile&v=2)
Und noch ein wenig deutlicher spezifiziert, was damit gemeint ist:
„Eine zeitgemäße regulatorische Vorgehensweise erfordert daher eine angemessene Interpretation und Umsetzung der bestehenden Arzneimittelgesetzgebung, mit dem Ziel, ein attraktives Forschungs- und Entwicklungsumfeld zu schaffen und zu bewahren, ohne dabei den oben genannten gesetzlichen Auftrag aus dem Auge zu verlieren.“
„Um derartige Voraussetzungen bieten zu können, muss eine Behörde den gleichen Sachverstand aufweisen wie ein Impfstoffhersteller. Sie muss sich darüber hinaus international so positionieren, dass sie alle wissenschaftlichen und gesetzlichen Vorgaben zur Prüfung und Zulassung von Impfstoffen im Wesentlichen mitbestimmt.“
Indem sie Paper mit jenen Firmen schreibt, die eine solche gesonderte Zulassung benötigen.
Zum Beispiel diese Publikation in Kooperation mit BioNTech und GSK:
Hinz T, Kallen K, Britten CM, Flamion B, Granzer U, Hoos A, Huber C, Khleif S, Kreiter S, Rammensee HG, Sahin U, Singh-Jasuja H, Türeci Ö, Kalinke U. The European Regulatory Environment of RNA-Based Vaccines. Methods Mol Biol. 2017;1499:203-222. doi: 10.1007/978-1-4939-6481-9_13. PMID: 27987152. https://pubmed.ncbi.nlm.nih.gov/27987152/
Wer Thomas Hinz ist und eine eingehende Analyse dieser PEI/BioNTech/TRON/GSK- Publikation findet man hier: https://www.corodok.de/wie-u-sahin/.
Thomas Hinz Fachgebiet sind therapeutische Impfstoffe. 2019 gab er im PEI eine Fortbildung zum Thema „Update on cancer vaccines“, was ganz zufällig das Gebiet ist, auf dem BioNTech hauptsächlich aktiv war/ist (https://www.pei.de/SharedDocs/Downloads/DE/newsroom/veranstaltungen/2019/regulatorische-weiterbildung-termine.pdf?__blob=publicationFile&v=2).
Im Organigramm findet man Hinz hier:

Er gehört wohl zum Prüflabor, dass die Chargen (nicht) geprüft hat.
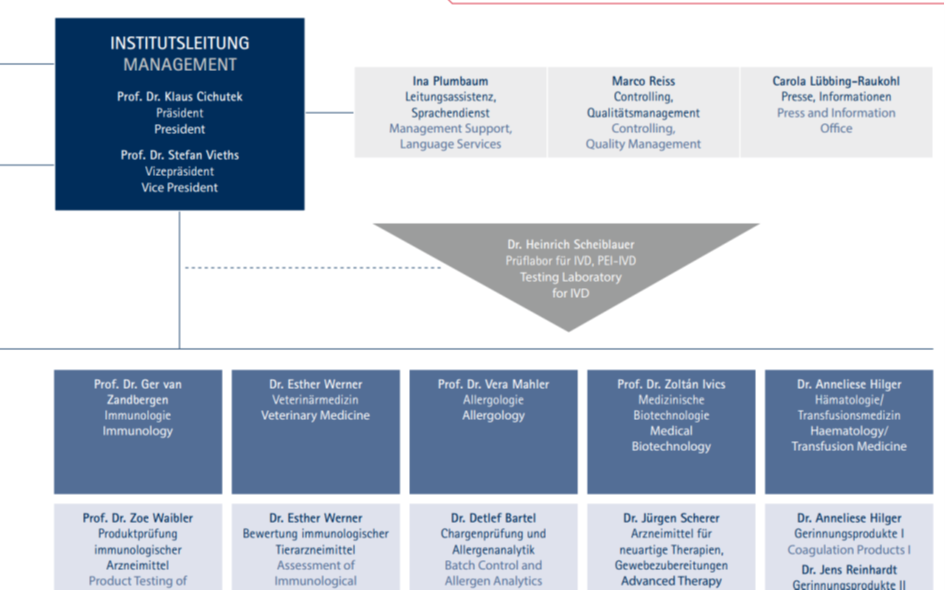
Unter Balance zwischen möglichen regulatorischen Vereinfachungen und notwendigen Anforderungen zu schaffen“ versteht das PEI anscheinend, mit diesen Firmen die entsprechenden Regeln so zu gestalten, wie es den Firmen am genehmsten ist.
EN: vaccine products are subject to approval, in Germany only by the Paul-Ehrlich-Institut, in all European Member States by the European Commission
DE: Impfstoffprodukte sind zulassungspflichtig, in Deutschland nur durch das Paul-Ehrlich-Institut, in allen europäischen Mitgliedsstaaten durch die Europäische Kommission
Das verstehe ich nicht so genau. Bei allen außer Deutschland, entscheidet die EU Kommission, nur in Deutschland das PEI? Oder muss das PEI noch zusätzlich zur EU-Kommission entscheiden?
2. Qualitätsbezogene regulatorische Anforderungen an Impfstoffe
EN: “As for common vaccines, the establishment of a fully quality-assured manufacturing process is of fundamental importance for COVID-19 vaccine development. This requires detailed process-specific developments and specifications as well as the implementation of suitable control measures including in-process controls. The entire production of vaccines must meet the requirements of “Good Manufacturing Practice” (GMP)”
DE: "Wie bei herkömmlichen Impfstoffen ist auch bei der Entwicklung des COVID-19-Impfstoffs die Etablierung eines vollständig qualitätsgesicherten Herstellungsprozesses von grundlegender Bedeutung. Dies erfordert detaillierte prozessspezifische Entwicklungen und Spezifikationen sowie die Durchführung geeigneter Kontrollmaßnahmen einschließlich prozessbegleitender Kontrollen. Die gesamte Produktion von Impfstoffen muss den Anforderungen der "Good Manufacturing Practice" (GMP) entsprechen.“
Wer das genau wissen will, kann das hier nachlesen:
https://drbine.substack.com/p/die-ema-wei-dass-der-dnase-i-verdau
Das Ganze lässt sich aber mit einem einzigen Bild abhandeln:

https://mega.nz/folder/WQo2QDaa#xp50i-ducJCz8cCHtq2AFg
Also der DNase I Verdau hat es wohl nicht geschafft mit GMP. Was nun PEI? Vor allem, weil das PEI an dieser Stelle die EU GMP Regeln zitiert, bei welcher BioNTech/Pfizer wohl nach exakt deren Regeln durchgefallen ist.
European Commission. COMMISSION DIRECTIVE 2003/94/EC of 8 October 2003: Laying down the Principles and Guidelines of Good Manufacturing Practice in Respect of Medicinal Products for Human Use and Investiga-Tional Medicinal Products for Human Use. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32003L0094 (accessed on 27 May 2021)
EN: “In Germany, the state authority in whose regional area the manufacturing facility is located grants this permit in consultation with the Paul-Ehrlich-Institut. For this purpose, the consistency of production is typically proven through three consecutive production processes for an identical product. A certain degree of flexibility has been applied for COVID-19 vaccines as regards the manufacturing range of these process performance qualification lots or by taking into account the respective data for similar products of the same platform technology”
DE: "In Deutschland erteilt die Landesbehörde, in deren regionalem Bereich sich die Produktionsstätte befindet, diese Genehmigung in Abstimmung mit dem Paul-Ehrlich-Institut. Zu diesem Zweck wird die Beständigkeit der Produktion in der Regel durch drei aufeinanderfolgende Produktionsprozesse für ein identisches Produkt nachgewiesen. Für COVID-19-Impfstoffe wurde ein gewisses Maß an Flexibilität in Bezug auf den Herstellungsbereich dieser Lose zur Qualifizierung der Prozessleistung oder durch Berücksichtigung der entsprechenden Daten für ähnliche Produkte derselben Plattformtechnologie angewandt.“
Damit hat nicht nur das PEI den schwarzen Peter sondern auch alle Bundesländer, in welchen produziert wurde, bzw. das Bundesland, in welchen der DNase I -Verdau stattfindet. Das wäre Hessen, weil das in der Fabrik in Marburg durchgeführt wird (https://www.pharma-food.de/ausruestung/fluessig-prozesstechnik/biontech-pfizer-vom-reaktor-bis-zum-impfzentrum-349.html). Damit kann man den GMP-Verstoß auf Landesebene hochkochen lassen, ohne dass der Bund dabei eingeschaltet werden müsste. Und auf Landeseben gäbe es auch einen eindeutig Zuständigen, der das Marburg Werk GMP zertifiziert hat.
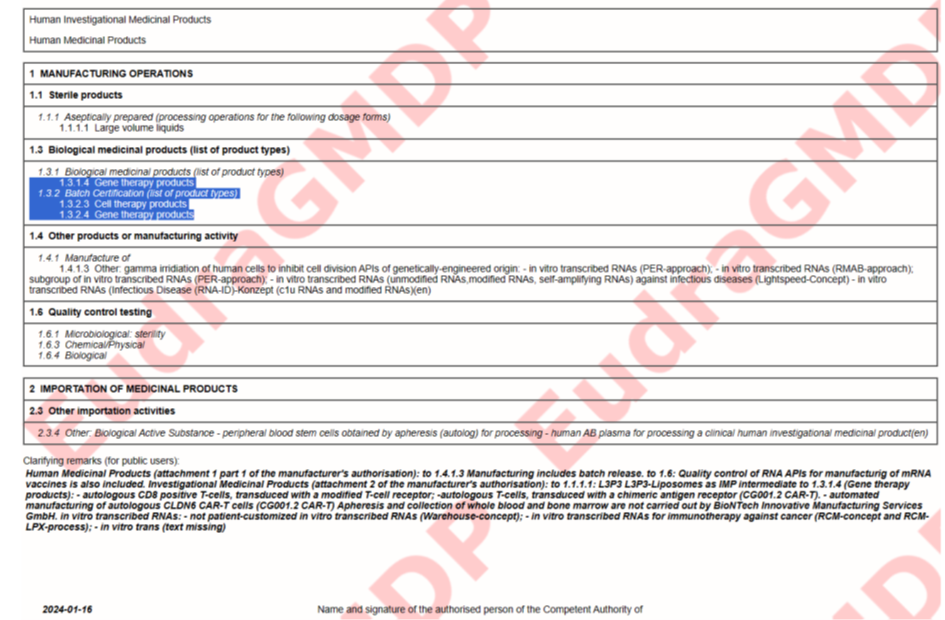
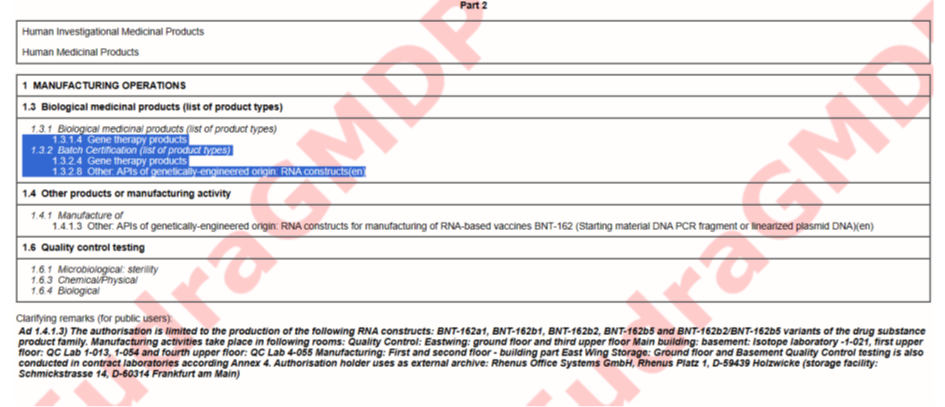
Schauen wir uns die GMP Zertifikate für verschiedene BioNTech Standunkte an Idar-Oberstein, Mainz und Marburg an (https://eudragmdp.ema.europa.eu/inspections/gmpc/searchGMPCompliance.do).
Schaut man sich die Zertifikate an, fällt auf:
Idar-Oberstein
Mainz, An der Goldgrube 12:
Mainz, Kupferbergterrasse 17
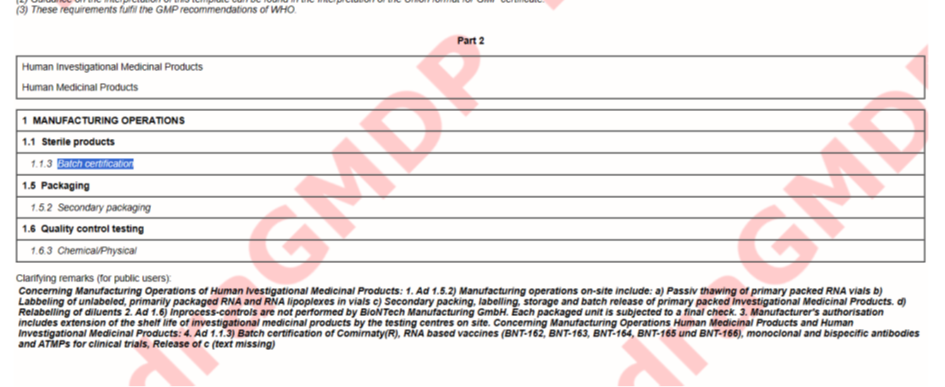
Marburg:

Die Studienmedikation wurde noch in Mainz hergestellt. Mainz hat jedoch eine GMP Zulassung für „gene therapy products“ also Gentherapieprodukte, genau wie Idar-Oberstein. Alle Produkte von BioNTech laufen unter „gene therapy products“, außer der Großproduktion von Comirnaty. Die Studienmedikation war also noch Gentherapie. Marburg läuft dann nur noch unter „Biological Medical Products“, obwohl das Gleiche hergestellt wird, wie in Mainz, nur in größerer Menge.
Gilt eine GMP Freigabe für „gene therapy products“ auch gleichzeitig für „Biological Medical Products“ oder fand die Herstellung der Studienmedikation nicht unter GMP statt?
Immerhin ist mit diesem GMP-Zertifikat klar, wer das durchgewunken hat: Das Regierungspräsidium Darmstadt. Wie praktisch, dass Langen, wo das PEI beheimatet ist, auch in Hessen liegt. Da hatten Sahin und Cichutek ja echt kurze Wege und konnten so einiges auf dem kleinen, persönlichen Dienstweg erledigen und direkt beim Regierungspräsidium Darmstadt vorbeischauen und da persönlich ein wenig Druck machen, damit das Werk in Marburg in Rekordzeit eine GMP-Zertifizierung bekam. Mir wurde von einem Vöglein geflüstert, dass das Regierungspräsidium Darmstadt schon vor der Plandemie sehr pharmafreundlich und hilfsbereit war.
ABER ganz ist die Bunderegierung laut „Project Lightspeed“ nicht raus.
…» Marett fügte hinzu, dass BioNTech jedoch über die Marburger Anlage einen Weg gefunden habe, der EU die Hälfte der 100-Millionen-Option in der ersten Jahreshälfte zur Verfügung stellen zu können, aber nur, wenn bürokratische Hindernisse überwunden werden könnten. Am Telefon erklärte er der Kommission, dass er «ihre Unterstützung brauche», um diesen Vorschlag umzusetzen. «Wir brauchen lediglich die Genehmigung unserer Produktionsanlage in Rekordzeit. Statt der üblichen sechs bis acht Monate müssen wir das in drei Monaten erledigt haben.» (Hervorhebung von mir)
Am 23. Dezember kam eine E-Mail, die die zusätzliche Bestellung bestätigte. Monate später würde die Produktionsstätte, für die BioNTech zunächst ohne finanzielle Unterstützung gekämpft hatte, der EU helfen, das Gesicht zu wahren. Das Bundesgesundheitsministerium in Berlin hatte mit lokalen Behörden in der Nähe von Marburg zusammengearbeitet, sodass bis Februar 2021 der Standort genehmigt und in Betrieb genommen werden konnte.“ (S. 308)
Die Genehmigung gab es dann direkt am 09. Februar 2021.
Jeder, der einmal eine GMP-Zertifizierung erlebt hat, kann da nur staunen, wie BioNTech das in nicht einmal einem Monat geschafft hat. Ich hatte im Referendariat mit dem Regierungspräsidium Freiburg zu tun, das war nur Kleinkram wegen Anerkennungen, das hat DEUTLICH länger gedauert und viele böse Mails gebraucht, bis jemand da gezuckt hat. Das zog sich fast 3 Semester bis kurz vor dem Referenariat. Da mussten sie dann den Arsch hoch bekommen.
Wie gut BioNTech sich mit GMP generell auskannte, und sehr erfahren und geübrt war, die nötigen Unterlagen für die Genehmigung eines riesigen Werkes zusammenzustellen. Die Details der GMP-Erfahrungen bei BioNTech findet man auch in „Project Lightspeed“:
„Ende 2008 hatte Kuhn in der Universitätsstadt Heidelberg einen Crashkurs in Good Manufacturing Practice (GMP) absolviert, um sich mit den weltweit anerkannten Richtlinien zur Qualitätssicherung bei der Herstellung von zugelassenen Arzneimitteln vertraut zu machen.“ (S. 245)
Kuhn hat also keine GMP-Erfahrung aus der Industrie, wo er das mal praktisch gelernt hätte, er hat einen Kurs gemacht. Kuhn, gehört aber zu Idar-Oberstein und nicht zu Marburg. Wer ist in Marburg zuständig? Lernt man in so einem Kurs auch, wie man ein Werk nach GMP zertifiziert? Welche Zertifizierungsstelle war darin involviert?
EN: “The quality documents contain detailed descriptions of the entire process steps with all relevant intermediate stages/products and complete information on the nature and origin of all raw materials. The quality requirements for vaccines are legally defined in Annex 1 to the European Directive 2001/83/EC.”
DE: “Die Qualitätsdokumente enthalten detaillierte Beschreibungen der gesamten Prozessschritte mit allen relevanten Zwischenstufen/Produkten und vollständige Informationen über die Art und Herkunft aller Rohstoffe. Die Qualitätsanforderungen für Impfstoffe sind in Anhang 1 der europäischen Richtlinie 2001/83/EG gesetzlich festgelegt.“
Damit sind wohl die Dokumente aus dem EMA-Leak gemeint: https://t.co/fFsopN1R00
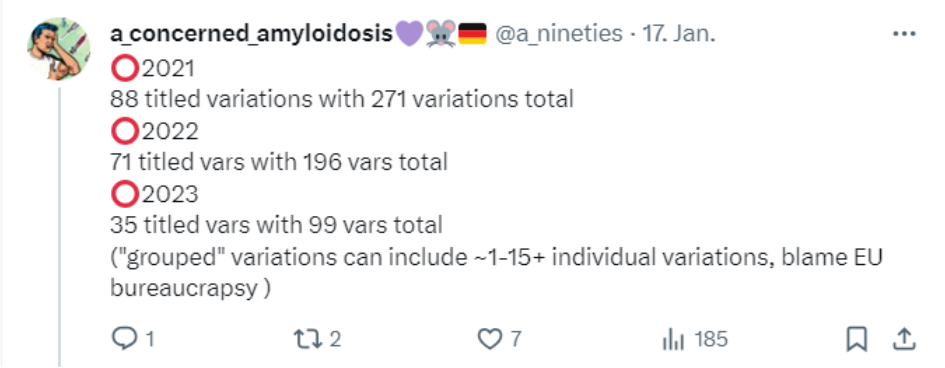
A-concerned_amyloidosis versucht irgendwie einen Überblick über die diversen Änderungen zu bekommen, die bei der EMA gelistet sind
(https://www.ema.europa.eu/en/documents/procedural-steps-after/comirnaty-epar-procedural-steps-taken-and-scientific-information-after-authorisation_en.pdf). Leider gibt es aktuell kein einziges, schön übersichtliches Dokument, in welchem der aktuelle Prozess selbst detailliert aufgeführt wurde, oder dieses Dokument ist mir einfach nicht bekannt. Die EMA-Leak Dokumente kommen dem noch am nächsten.
Der vom PEI zitierte EU-Link als Referenz für die Regeln, an die man sich angeblich gehalten hat, ist mittlerweile leider tot, aber es könnte sich um dieses Dokument handeln https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/directive-200183ec-european-parliament-and-council-6-november-2001-community-code-relating-medicinal-products-human-use_en.pdf (Danke an a_concerned_amyloidosis).

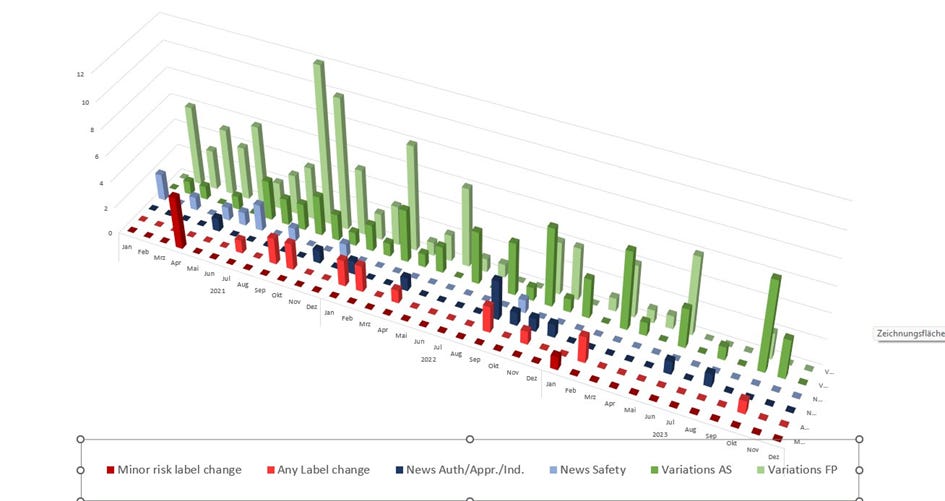
https://twitter.com/a_nineties/status/1749312065481617843/photo/1
Download der XLS-Datei auf MEGA (Danke!)
EN: “Furthermore, all implemented control processes including the specifications established for release testing, which were implemented to ensure the required and consistent vaccine quality, must be precisely described and justified. For licensure, the manufacturing process and the control methods used must be validated.”
DE: "Darüber hinaus müssen alle implementierten Kontrollprozesse einschließlich der für die Freigabeprüfung festgelegten Spezifikationen, die zur Sicherstellung der erforderlichen und gleichbleibenden Impfstoffqualität implementiert wurden, genau beschrieben und begründet werden. Für die Zulassung müssen der Herstellungsprozess und die verwendeten Kontrollmethoden validiert werden.“
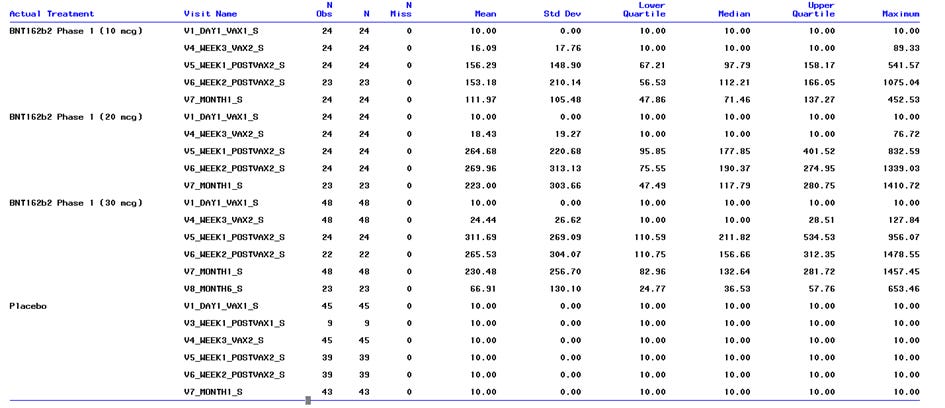
Die Suche nach „process validation“ im Abstractor für die Pfizer Dokumente (https://vaccines.shinyapps.io/abstractor/) bringt genau 0 Treffer. Aber nicht alle Dokumente scheinen maschinenlesbar zu sein. Ich kenn kein entsprechendes Dokument, was bei den Datenmengen nicht heißt, dass es die nicht gibt.
Aber schon der Vergleich von EU zu US Chargen sieht mir nicht nach validiert aus:

https://twitter.com/joshg99/status/1725140344436658649
EU bedeutet aber nicht BioNTech, das könnte „nur“ die Variation der Pfizer Produktionsanalgen sein und nicht einmal ein Vergleich zwischen Pfizer und BioNTech.

Die haben nicht einmal die Vergleichbarkeit von Prozess 1 und Prozess 2 validiert, weil die Daten der 250 Personen zwar erhoben aber nie ausgewertet wurden, weil das Produkt ja schon im Markt war.
https://www.nejm.org/doi/suppl/10.1056/NEJMoa2034577/suppl_file/nejmoa2034577_protocol.pdf

„Normalerweise können solche Änderungen durch analytische Daten gestützt werden, doch aufgrund der Zulassungslandschaft für COVID-19-Impfstoffe wurde im Oktober 2020 ein exploratives Ziel in die in der Studie C4591001 hinzugefügt, um die Sicherheit und Immunogenität von Impfstoffen zu beschreiben, die Prozess 1" oder "Prozess 2" hergestellten Impfstoffe bei Teilnehmern im Alter von 16 bis 55 Jahren zu beschreiben. Dieses wurde entfernt und in Protokolländerung 20 im September 2022 dokumentiert. September 2022 gestrichen und dokumentiert, da Impfstoffe, die nach dem "Verfahren 2" hergestellt werden, in großem Umfang verwendet werden. Daher wurde dieser Prozessvergleich nicht als Teil der formalen Dokumentation innerhalb der Protokolländerung durchgeführt.“
https://dailysceptic.org/wp-content/uploads/2023/09/IR-23-510.pdf
„Gleichbleibenden Impfstoffqualität“ sähe für mich irgendwie anders aus.
Aus den „für die Freigabeprüfung festgelegten Spezifikationen“ wird aktuell ein Staatsgeheimnis gemacht, besonders, was die Grenzwerte für DNA angeht, da winden sich sowohl das PEI als auch die SWISSMEDIC. Laut SWISSMEDIC gibt es da keine Grenzwerte, sondern nur freiwillige Zugeständnisse der Hersteller. Aktuell mauern alle in dieser Hinsicht.
Wenn die Kontrollmethoden validiert wären, wie kann es dann sein, dass man keinen Extinktionskoeffizienten für die modRNA bestimmt hat? Das hätte einem doch auffallen müssen, dass modRNA anders im UV-Spektrometer bei 260 nm absorbiert als biomRNA/uRNA.

„Wir berichten, dass N1-Methylpseudouridin bei 260 nm ca. 40 % weniger UV-Licht absorbiert als Uridin, und dass sein Einbau in mRNAs zu einer Unterschätzung der nukleosidmodifizierten mRNA-Konzentrationen führt, mit einem Fehler von 5-15 %, und zwar in Abhängigkeit von der mRNA-Sequenz.” (biorxiv.org/content/10.1101/2023.07.27.550903v2.full.pdf).
Es hätte auch auffallen müssen, dass qPCR kleine DNA-Fragmente unterschätzt und daher ungeeignet ist:

https://twitter.com/Patent_SUN/status/1747644965990359530 und mRNA-Impfstoffe - DNA in Covid-Impfstoffen: Nur unkontrollierte Messfehler? - Teil 2 | Cicero Online
Was meint das PEI also mit „alle implementierten Kontrollprozesse einschließlich der für die Freigabeprüfung festgelegten Spezifikationen“? Man kann diese Spezifikationen gar nicht messen, weil es da Interferenzen durch die LNP-Formulierung gibt. Das stellt aktuell sowohl Kritiker der modRNA-Produkte als auch Verfechter vor ein experimentelles Problem.
EN: “These tests also include controls and the minimization of any possible contamination.”
DE: “Zu diesen Tests gehören auch Kontrollen und die Minimierung möglicher Kontaminationen.“
Minimierung bedeutet nicht Eliminierung. Hier wären wir beim Problem der DNA-Verunreinigungen, die bereits angesprochen wurden. Die wurden so gut es geht minimiert, wie es aussieht. Ganz weg geht nicht, also ist man zufrieden, mit dem, was man erreicht hat.
Nur, was genau wurde minimiert? Den Firmen ist die DNA-Verunreinigung egal, da kräht keine Behörde danach. Was minimiert wird ist die dsRNA-Verunreinigun, die kaum einer auf dem Schirm hat. Patent_Sun hat diesbezüglich noch ein Patent gefunden. Man optimiert die modRNA-Ausbreute, minimiert die dsRNA und erhöht gleichzeitig aber MASSIV die DNA-Kontamination. Und das wurde wohl durchgewunken, weil BioNTech, das das EMA einfach verschwiegen hat (aber so dumm war, es ins Patent zu schreiben).

Maria Gutschi erklärt das Patent dann ein wenig mehr von der chemischen Seite:
EN: “For the end product, it must be ensured that the active ingredient content is constant and that a homogeneous formulation of the end product is guaranteed. All other quality-determining parameters must also be checked and confirmed. This is ensured by intensive testing of the end product (prior to the official release) by the manufacturer in accordance with the test program established as part of the approval process. Approved vaccines are also subject to batch testing by the Paul-Ehrlich-Institut or another official control laboratory from the European OMCL network.”
DE: „Für das Endprodukt muss sichergestellt sein, dass der Wirkstoffgehalt konstant ist und eine homogene Formulierung des Endprodukts gewährleistet ist. Auch alle anderen qualitätsbestimmenden Parameter müssen überprüft und bestätigt werden. Dies wird durch eine intensive Prüfung des Endprodukts (vor der offiziellen Freigabe) durch den Hersteller nach dem im Rahmen des Zulassungsverfahrens festgelegten Prüfprogramm sichergestellt. Zugelassene Impfstoffe unterliegen zudem einer Chargenprüfung durch das Paul-Ehrlich-Institut oder ein anderes amtliches Kontrolllabor aus dem europäischen OMCL-Netzwerk.“
Über diese Diskussion und was das PEI testet oder nicht testet, habe ich bereits einen ganzen Artikel geschrieben.
https://drbine.substack.com/p/wie-man-sich-aus-einen-eigenen-worten
Mal behauptet das PEI vor Gericht nur Sichtprüfung zu machen, ein anderes Mal behauptet das PEI im Schichtdienst nach ISO geprüft zu haben. Bei den Spezifikationen wird gemauert, Prüfprotokolle will man nicht rausrücken:
„[…] Aufgrund unserer bisherigen Erfahrungen mit IFG-Anträgen auf Einsicht in Prüfprotokolle und -ergebnisse von Chargenprüfungen machen wir Sie darauf aufmerksam, dass davon auszugehen ist, dass die konkreten Testbeschreibungen und -ergebnisse in diesen Prüfprotokollen als Betriebs und Geschäftsgeheimnisse einzustufen sind und daher geschwärzt werden müssen. Es ist daher wahrscheinlich, dass Sie diesen Unterlagen kaum für Sie relevante Informationen entnehmen können.” https://individuelle-impfentscheidung.de/fileadmin/PDF/PEI_Antwort_geschw.pdf
Das wirft natürlich Fragen auf, was es zu verbergen gibt, wenn doch alles vollkommen sauber und nach ISO-und GMP-Normen gelaufen ist und stärkt nicht gerade das Vertrauen in das PEI und die SWISSMEDIC, die in ihren Antworten entweder beim PEI abgeschrieben hat, oder man hat einen gemeinsam genutzten Antwortenkatalog.
EN: “Nevertheless, before entering the clinical study phase, it must be ensured that the clinical trial material is sufficiently characterized and has been produced using a defined and consistent manufacturing process. This is examined in detail by the Paul-Ehrlich-Institut as part of the approval procedure for the clinical trial.”
DE: „Dennoch muss vor dem Eintritt in die klinische Studienphase sichergestellt werden, dass das klinische Prüfmaterial ausreichend charakterisiert ist und nach einem definierten und einheitlichen Herstellungsprozess hergestellt wurde. Dies wird im Rahmen des Genehmigungsverfahrens für die klinische Prüfung durch das Paul-Ehrlich-Institut eingehend geprüft.“
Ich bin bereits in diesem Artikel darauf eingegangen, dass man keine Ahnung hat, wie das Produkt überhaupt funktioniert. Dass Toxikologische und Pharmakologische Studien ausgelassen wurden.
Man weiß nicht einmal, ob und wie das Produkt in der Leber abgebaut wird. Es gibt nur einen vorgeschlagenen Abbauweg, den man aber nicht experimentell überprüft oder ansatzweise untersucht hat (https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf S.46).
Die Struktur der LNPs war unbekannt. Man ging davon aus, dass es schon irgendwie wie Onpattro aussehen würde, was sich als vollkommen falsch erwiesen hat. Das Produkt sieht keinem bisher bekannten LNP ähnlich und hat NICHTs aber auch gar NICHTS mit den eingereichten Schemazeichnungen zu tun. Das habe ich bereits in diesem Substack ausführlich behandelt:
https://drbine.substack.com/p/die-plorre-ist-wohl-innen-flussig
Das es bis heute keinen definierten Produktionsprozess gibt, habe ich auch bereits behandelt.
„Dies wird im Rahmen des Genehmigungsverfahrens für die klinische Prüfung durch das Paul-Ehrlich-Institut eingehend geprüft.“
WAS hat das PEI da eigentlich geprüft?! Wenn die das geprüft haben, müssen sie doch wissen, dass der Satz davor eine einzige, große Lüge ist.
3. Anforderungen für die nicht-klinische Prüfung
EN: “As part of these investigations, studies of the primary pharmacological effects have to be carried out, on the basis of which vaccine-specific dose–effect relationships (pharmacodynamics) can be identified and defined. This is one of the bases for the development of the first indications for dose finding and the establishment of a suitable vaccination schedule for later use in humans. In contrast to traditional drugs, which are applied repeatedly and over long periods of time, typically no data on pharmacokinetics, accumulation, and biodistribution have to be collected for vaccines, which are usually administered with few doses and relatively small amounts of substance. However, in the case of live attenuated and replication-competent vector vaccines or for totally novel platform technologies and adjuvants, investigations on the possible distribution and persistence in the body and on the excretion profile (“shedding”) need to be carried out. On a case-by-case regulatory evaluation, such studies have also been required for certain COVID-19 vaccines.“
DE: "Im Rahmen dieser Untersuchungen müssen Studien zu den primären pharmakologischen Wirkungen durchgeführt werden, auf deren Grundlage impfstoffspezifische Dosis-Wirkungs-Beziehungen (Pharmakodynamik) ermittelt und definiert werden können. Dies ist eine der Grundlagen für die Entwicklung der ersten Indikationen für die Dosisfindung und die Festlegung eines geeigneten Impfschemas für die spätere Anwendung beim Menschen. Im Gegensatz zu herkömmlichen Arzneimitteln, die wiederholt und über lange Zeiträume angewendet werden, müssen in der Regel keine Daten zur Pharmakokinetik, Akkumulation und Biodistribution gesammelt werden, da Impfstoffe in der Regel mit wenigen Dosen und relativ kleinen Substanzmengen verabreicht werden. Im Falle von abgeschwächten Lebendimpfstoffen und replikationskompetenten Vektorimpfstoffen oder für völlig neue Plattformtechnologien und Adjuvantien Untersuchungen zur möglichen Verteilung und Persistenz im Körper und zum Ausscheidungsprofil ("Shedding") durchgeführt werden müssen. Bei einer fallweisen regulatorischen wurden solche Studien auch für bestimmte COVID-19-Impfstoffe gefordert.“
Ich muss mich hier wohl wiederholen, langsam komme ich mir bei einigen Dokumenten vor wie eine tibetanische Gebetsmühle.

„Der exakte immunologische Mechanismus, der für Schutz gegen SARS-COV-2 verantwortlich ist, ist unbekannt“( https://www.fda.gov/media/151733/download (S. 15))

Und Obwohl man wusste, dass 30µg nicht besser sind als 20µg und nur mehr Nebenwirkungen machen, hat man sich wohl einvernehmlich auf 30µg geeinigt.
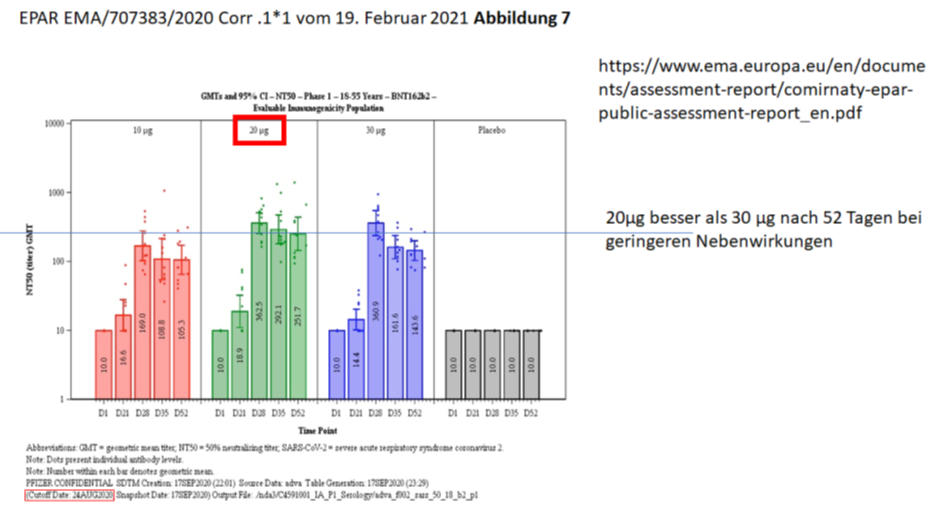



Obwohl man beim PEI eigentlich weiß: „Anders als bei therapeutischen Arzneimittel steigt die Wirkung eines Impfstoffes, d. h. die Auslösung einer Immunantwort, in der Regel jedoch nicht parallel zur Erhöhung der Dosis“ (https://www.pei.de/SharedDocs/Downloads/wiss-publikationen-volltext/bundesgesundheitsblatt/2015/2015-zulassung-impfstoffe-empfehlung-stiko-kriterien-nutzen-risiko.pdf?__blob=publicationFile&v=2)
Und daher hat man Dosisfindungen mit komplett anderen Konstrukten durchgeführt, als letztendlich eingesetzt wurden.
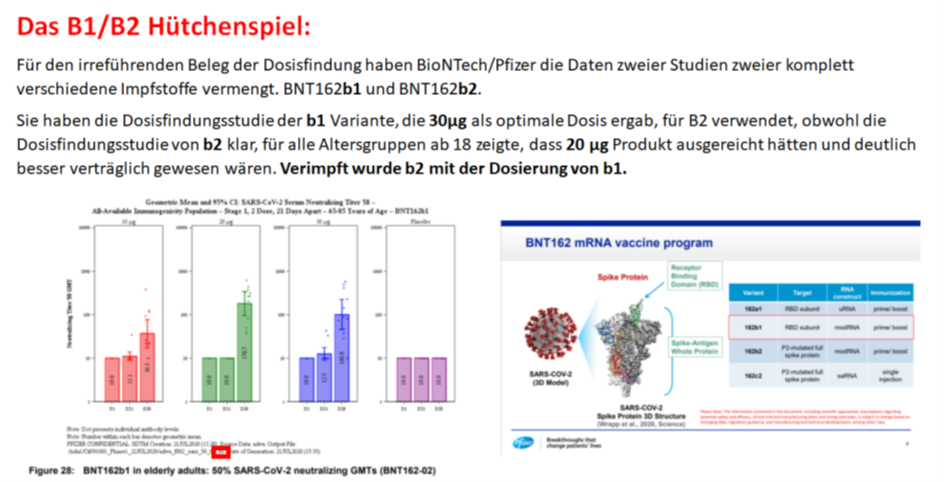
Des Weiteren wurden die klinische Phase 3 Studie begonnen, bevor die ersten Mausstudien überhaupt abgeschlossen waren, also ohne irgendwelche Daten für die optimale Dosierung von Menschen hätten vorliegen können. In diesem Bereich ist a_concerned_amyloidosis auf Twitter der Experte:

https://twitter.com/a_nineties/status/1752029173819060370/photo/1
Was Shedding angeht, da gibt es zwar ein Pfizerdokument wo dies erwähnt wird, aber es wurden keine weiteren Studien diesbezüglich aufgesetzt.

- Ein männlicher Teilnehmer, der an der Studie teilnimmt oder sie abgebrochen hat, setzt eine eine Partnerin vor oder um den Zeitpunkt der Empfängnis.
- Bei einer Frau wird eine Schwangerschaft festgestellt, während sie aufgrund einer Umweltexposition der Studienintervention aufgrund einer Umweltexposition schwanger ist. Im Folgenden sind Beispiele für Umweltexposition Exposition während der Schwangerschaft:
- Ein weibliches Familienmitglied oder ein Gesundheitsdienstleister berichtet, dass sie schwanger ist, nachdem sie nachdem sie der Studienintervention durch Einatmen oder Hautkontakt ausgesetzt war.
- Ein männliches Familienmitglied oder ein medizinischer Betreuer, der der Studienintervention der Studienintervention durch Inhalation oder Hautkontakt ausgesetzt war, exponiert seine Partnerin vor oder um den Zeitpunkt der Empfängnis herum.
Die FDA definiert Shedding übrigens wie folgt:
„Im Sinne dieses Leitfadens bedeutet der Begriff "Shedding" die Freisetzung von virus- oder bakterienbasierte Gentherapie-Produkten (VBGT) oder onkolytischen Produkten aus dem Patienten auf einem oder allen der folgenden Wege: Ausscheidungen (Kot), Sekrete (Urin, Speichel, Nasopharyngealflüssigkeit usw.) oder über die Haut (Pusteln, Wunden). Shedding unterscheidet sich von der Biodistribution, da letztere beschreibt, wie ein Produkt vom Verabreichungsort im Körper des Patienten verteilt wird, während ersteres beschreibt, wie es Ausscheidung oder Freisetzung aus dem Körper des Patienten. Beim Shedding besteht die Möglichkeit der Übertragung von VBGT oder onkolytischer Produkte von behandelten auf unbehandelte Personen (z. B. enge Kontakte und Angehörige der Gesundheitsberufe).“ https://www.fda.gov/media/89036/download
So wie sich das liest hätte man das definitiv für die Adenovirenprodukte machen müssen, denn die bisher zugelassenen Adenoviren Gentherapien shedden alle:
Ein Beispiel wäre Luxturna:


https://www.ema.europa.eu/en/documents/product-information/luxturna-epar-product-information_en.pdf
Weitere Beispiele wären Roctavian (https://www.fda.gov/media/170455/download) und Zolgensma (https://www.fda.gov/media/126109/download).
Also zumindest für Johnson&Johnsons Produkt und für das AstraZeneca Produkt hätte man Shedding-Studien durchführen müssen.
Was die „möglichen Verteilung und Persistenz im Körper“ angeht, hat das PEI sich nicht gerade mit Ruhm bekleckert. Man hat nicht von Anfang an klar gestellt, dass sich das Produkt im ganzen Körper verteilt, obwohl die Verteilungsstudien der Hersteller das klar gezeigt haben (https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf S. 45).
Was die Persistenz des Produktes im Körper angeht, so ergeben sich einige Probleme:
1. Wie kann man die Lipide im Körper nachweisen. Mir ist kein chemisch-eindeutiger Beweis bekannt. Wie hat man das also in den Studien gemacht? Wie hat man ausgeschlossen, dass die Lipide sich nicht in die Zellemembranen einbauen und diese stören?
2. Hat man die Verweildauer der modRNA in den Zellen gemessen? Mir sind keine Daten der Hersteller bekannt. Erst Jahre später kamen unabhängige Daten von Forschergruppen rein.
3. Hat man untersucht, wo und wie das N1-Methylpseudouridin (m1Ψ) abgebaut oder recycelt wird?
4. Zählt das Spike-Protein als Produkt? DAS persistiert scheints ziemlich lange.
Diese Daten sind mir bisher nicht bekannt. Ich vermute, es gibt diese Daten nicht. Wie konnte das PEI diese Daten dann beurteilen? Auch ist der Abbauweg der LNPs, wie bereits erwähnt, selbst dem Hersteller BioNTech/Pfizer aktuell unbekannt. Es gibt nur einen vorgeschlagenen metabolischen Abbauweg, der nie bewiesen wurde.
Immerhin, eine „reapeat-dose-toxicity“ Studie in Ratten gab es:

https://phmpt.org/wp-content/uploads/2023/01/125742_S16_M1_psp-track.pdf#page=14
Meines Wissens muss so eine Studie aber eine Dosis mehr, als für den Menschen vorgesehen umfassen. Ich glaube nicht, dass man bis 5 oder 7 Dosen probiert hat.
Besonders lange hat man die Ratten nicht beobachtet. Ob diese Tiere wohl noch leben würden, hätte man sie langfristig beobachtet?
Bei den Tierstudien habe ich aber, zugegebenermaßen, noch große Wissenslücken, da ich selbst nie Tierversuche gemacht habe und daher nicht vertraut bin mit den Auswertungen und der Vorgehensweise.
EN: “and extensive histological examinations of organ systems in order to reliably identify possible intolerance indicators. If approval for pregnant women is sought, studies of embryo–fetal toxicityare required.”
DE: "und umfangreiche histologische Untersuchungen von Organsystemen, um mögliche Unverträglichkeitsindikatoren zuverlässig zu identifizieren. Wird eine Zulassung für Schwangere angestrebt, sind Untersuchungen zur embryofetalen Toxizität erforderlich".
Dazu gibt es von mir einen laaaaaaaangen Artikel:
https://drbine.substack.com/p/pazentagate-revisited
Oder, um es mit den Worten von Jelena Vojicic, der medizinischen Leiterin für Impfstoffe bei Pfizer Canada, in einer E-Mail von 2022 zu sagen
„Die Aufnahme in die Studie wurde mit unvollständigen Zahlen gestoppt, weil die Rekrutierung langsam war und es unvernünftig/unangemessen wurde, schwangere Frauen nach dem Zufallsprinzip auf Placebo zu setzen, angesichts der Menge an Beobachtungsdaten, die belegen, dass der Impfstoff sicher und wirksam ist, und angesichts der zunehmenden Zahl von Fachausschüssen, die die Immunisierung schwangerer Frauen unterstützen“ https://www.theepochtimes.com/health/pfizer-says-it-ended-covid-19-vaccine-pregnancy-trial-early_5088375.html?utm_source=partner&utm_campaign=ZeroHedge&src_src=partner&src_cmp=ZeroHedge
Die Abortrate war SEHR hoch.
Was die histologischen Untersuchungen angeht, die kamen letztendlich von Arne Burkhardt und das Spike wurde dabei in der Placenta nachgewiesen.
Es gibt dazu auch mittlerweile wissenschaftliche Publikationen, die zu einem vernichtenden Urteil bezüglich der modRNA-Produkte kommen:
“Wenn man sie auf die verfügbare Zeit, die verabreichten Dosen oder die Anzahl Anzahl der geimpften Personen, übersteigen alle COVID-19-Impfstoff-AEs bei weitem das Sicherheitssignal bei allen anerkannten Schwellenwerten. Diese Ergebnisse erfordern ein weltweites Moratorium für die Anwendung von COVID-19-Impfstoffen in der Schwangerschaft. […] Die Verabreichung von COVID-19-Impfstoffen in der Schwangerschaft und bei Frauen Frauen im reproduktiven Alter sollte sofort gestoppt werden, bis diese Sicherheitssignale vollständig untersucht werden können.” (https://jpands.org/vol28no1/thorp.pdf).
EN: Furthermore, depending on the type of vaccine and the planned indication and application, reproductive, genotoxicity, or carcinogenicity studies may be necessary.
DE: „Außerdem können je nach Art des Impfstoffs und der geplanten Indikation und Anwendung Reproduktions-, Genotoxizitäts- oder Karzinogenitätsstudien erforderlich sein.“
Können erforderlich sein ist ja ein Konditionalsatz, zumindest teilweise, denn die Art des Produktes, die eigentlich Einschränkung, wird in diesem Satz nicht definiert oder festgelegt. Für modRNA-Produkte waren diese Art Studien ganz offensichtlich nicht notwendig.


https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf
Dass Reproduktionstoxizitätsdaten bei einer Generationendauer beim Menschen von Minimum 12 Jahren nicht vorliegen können, versteht sich von selbst. Die sinkenden Geburtenraten in diversen Ländern lassen zwar schlimmes ahnen, aber da in diesen Ländern meist auch die Heiratsraten und die Wirtschaftsdaten in den Keller gingen, ist eine eindeutige Aussage aktuell noch nicht absolut eindeutig zu treffen. Zu viele Faktoren, die hineinspielen könnten, da streiten sich die Statistiker noch. Ein Trend rückläufiger Geburtenraten ist ersichtlich, aber auch die politisch-ökonomische Lage, teils durch die Covid-Maßnahmen mit verursacht, ist eben auch nicht unerheblich. Wenn man nicht weiß, ob man im nächsten Monat noch einen Job hat oder die Heizkosten zahlen kann, wird man keine Kinder bekommen.
EN: Since wild-type (wt) mice are not permissive to SARS-CoV-2 infection, huACE2-transgenic mice are used for COVID-19 vaccine-related studies in the mouse model, which express the human ACE2 receptor on cells and are therefore readily infected with SARS-CoV-2 and develop a clear disease phenotype.
DE: Da Wildtyp-Mäuse (wt) keine SARS-CoV-2-Infektion zulassen, werden für COVID-19-impfstoffbezogene Studien im Mausmodell huACE2-transgene Mäuse verwendet, die den menschlichen ACE2-Rezeptor auf Zellen exprimieren und daher leicht mit SARS-CoV-2 infiziert werden und einen eindeutigen Krankheitsphänotyp entwickeln.
In den Unterlagen habe ich bisher aber nur von BALB/c Mäusen (z. Bsp hier https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M2_24_nonclinical-overview.pdf) gelesen. Der Abstractor bringt 27 Treffer für BALB/c . Des Weiteren habe ich von Wistar Han Ratten (z. Bsp. hier https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M1_priority-review-request-1.pdf) gelesen, der Abstractor hat 324 Treffer für diesen Laborrattentyp.
Die Suche mit dem Abstractor nach huACE2 bringt 0 Treffer.
Die Suche mit dem Abstractor nach humanized, bringt nur einen Treffer bezüglich eines humanisierten Antikörpers in anderem Zusammenhang.
Man hat das Produkt, bzw. das daraus resultierende Spike-Protein, also nicht auf seine Reaktion mit humanisiertem huACE2 in Nagern getestet, sondern auf Nager ACE2. Und dann wundert man sich, wenn man keine Nebenwirkungen bemerkt hat.
Laut PEI hat man humanisierte Mäuse und Ratten genommen, was falsch/eine Lüge ist.
Eine übersichtliche Auflistung der Tierversuchstiere findet man in australischen Daten (foi-2389-03-1.pdf (tga.gov.au)):


Von humanisiert, steht da jedoch nichts.
Dieses Paper fasst die Tierversuche sehr schön übersichtlich zusammen (https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.2c00010). Auch hier, kein Hinweis auf humanisierte Versuchstiere.

EN: For all COVID-19 vaccine candidates, relevant data on the induced immune response (e.g., antibody titers, T cell responses, cytokines) must be collected for the intended application regimen (dose strength, number and timing of administration) in a suitable animal model before entering human clinical trials. These studies serve as the basis for the vaccination regimens analyzed in subsequent clinical trials. With regard to preclinical safety trials, the specific requirements are dependent on the product. For COVID-19 vaccine products utilizing established platform technologies of DNA, RNA, or vector vaccines, and for which relevant safety data from animal experiments (in particular, repeat-dose toxicity) for similar platform vaccines are available (e.g., vaccine design utilizes an established technology platform but contains different antigens and is targeted against other infectious diseases), essential parts of pre-clinical testing can take place in parallel to the clinical I/IIa trial. For entirely novel vaccine candidates, the full pre-clinical test program is mandatory before entering clinical phase I.
DE: Für alle COVID-19-Impfstoffkandidaten müssen relevante Daten zur induzierten Immunantwort (z. B. Antikörpertiter, T-Zell-Reaktionen, Zytokine) für das vorgesehene Anwendungsschema (Dosisstärke, Anzahl und Zeitpunkt der Verabreichung) in einem geeigneten Tiermodell erhoben werden, bevor sie in klinische Studien am Menschen eintreten. Diese Studien dienen als Grundlage für die Impfschemata, die in den anschließenden klinischen Studien analysiert werden. Die spezifischen Anforderungen an die präklinischen Sicherheitsstudien hängen vom jeweiligen Produkt ab. Für COVID-19-Impfstoffprodukte, die etablierte Plattformtechnologien von DNA-, RNA- oder Vektorimpfstoffen nutzen und für die relevante Sicherheitsdaten aus Tierversuchen (insbesondere Toxizität bei wiederholter Verabreichung) für ähnliche Plattformimpfstoffe vorliegen (z. B. Impfstoffdesign nutzt eine etablierte Technologieplattform, enthält aber andere Antigene und ist gegen andere Infektionskrankheiten gerichtet), können wesentliche Teile der präklinischen Tests parallel zur klinischen I/IIa-Studie durchgeführt werden. Für völlig neuartige Impfstoffkandidaten ist das vollständige präklinische Testprogramm obligatorisch, bevor die klinische Phase I beginnt.
Relevante Daten sind schwer zu erheben, wenn man nicht weiß, was relevant ist, weil man nicht weiß, wie das Produkt überhaupt funktioniert. Das hatte ich ja bereits oben ausgeführt.
In den Worten von BioNTech, aus dem MAI 2021 ausgedrückt, steht man vor folgendem Problem:
“Herkömmliche Sicherheitsstudien können zwar starke Beeinträchtigungen des Immunsystems aufdecken, liefern aber keine Informationen über moderate und geringe Immuntoxizität. Daher sind zusätzliche Studien erforderlich, die sich mit der Sekretion von Zytokinen und Chemokinen, Anti-Drug-Antikörpern (ADAs) sowie der Komplementaktivierung befassen, um potenzielle Sicherheitsbedenken wie Immunaktivierung oder -beeinträchtigungen vorherzusagen. […] Während diese Sicherheitsstudien begrenzte Daten zur Immunaktivierung durch angewandte LNPmRNA-Medikamente liefern, besteht ein eindeutiger Bedarf an zusätzlichen Daten, die sich mit folgenden Themen befassen würden: Vergleich von Modellsystemen und Assays sowie Festlegung optimaler prädiktiver Panels, neuer Biomarker und optimaler Testzeit. Darüber hinaus würde ein besseres Verständnis der Bereiche und Grenzwerte für präklinische Zytokin-/Chemokin-Messungen und die Ermittlung von Unterschieden zwischen menschlichen Spendern den prädiktiven Wert präklinischer Studien für die Klinik erhöhen.“ (https://pubmed.ncbi.nlm.nih.gov/34068715/)
Im Kartext: Wir haben auch Monate, nachdem bereits Milliarden von Menschen ein solches Produkt erhalten haben (das aber als Impfung deklariert ist und daher gilt das, was da steht natürlich juristisch nicht), keine Ahnung wie man das Zeugst auf Toxizität prüfen müsste, weil es noch keine Essays und Standards dafür gibt.“
Was das geeignete Tiermodell angeht, währen das humanisierte Nager gewesen. Die FDA verlang Zellkultur und gemacht wurden, neben den bereits erwähnten nicht humanisierten Nagern, vollkommen ungeeignete Primatenversuche und das hat man so auch in die Publikationen reingeschrieben:

„Wir kommen zu dem Schluss, dass das 2-4-jährige Männchen-Makaken-Herausforderungsmodell in erster Linie ein Modell für eine SARS-CoV-2-Infektion und nicht für eine COVID-19-Erkrankung ist.“ (https://www.nature.com/articles/s41586-021-03275-y). Im englischen Original mit einem Rechtschreib- und einem Grammatikfehler in diesem entscheidenden Satz.
Man hatte also Nagermodelle, die nicht aussagekräftig waren.
Man hatte also Primatenmodelle, die nicht aussagekräftig waren.
Das war aber egal, denn man hat das Ergebnis der Tierversuche gar nicht abgewartet, bevor man es am Menschen ausprobierte. Obwohl es sich um völlig neuartige Impfstoffkandidaten handelte und das vollständige präklinische Testprogramm obligatorisch gewesen wäre, bevor die klinische Phase I begann. Und das muss das PEI zu Zeitpunkt des Erscheinens ihres Artikels gewusst haben. Die mussten die Daten kennen und haben eiskalt gelogen.

https://twitter.com/a_nineties/status/1752028073959047620
Leider hat a_conderned_amyloidosis dazu noch keinen Substack geschriebe, hat aber freundlicherweise sein XLS Sheet mit den Studiennummern und Daten gepostet.
EN: An additional complication that was observed in animal experiments with SARS-CoV-1 and MERS vaccines was enhanced respiratory disease (ERD). Various factors are thought to contribute to the occurrence of ERD. These include a shifted profile of T-helper cell (Th) responses towards an enhanced Th2 profile, characterized by the increased expression of various cytokines (e.g. IL-4, IL-5, IL-6, IL-9, IL- 13, and IL-17E (IL-25)), the immigration of eosinophilic cells, and the reprogramming of tissue macrophages in the lungs from a regenerative phenotype to a proinflammatory phenotype, associated with an increase in IL-1, IL-6, IL-8, CXCL-10, and MCP-1.
DE: Eine zusätzliche Komplikation, die in Tierversuchen mit SARS-CoV-1- und MERS-Impfstoffen beobachtet wurde, war die verstärkte Atemwegserkrankung (ERD). Es wird angenommen, dass verschiedene Faktoren zum Auftreten von ERD beitragen. Dazu gehören eine Verschiebung des Profils der T-Helferzellen (Th) hin zu einem verstärkten Th2-Profil, das durch die verstärkte Expression verschiedener Zytokine (z. B. IL-4, IL-5, IL-6, IL-9, IL-13 und IL-17E (IL-25)) gekennzeichnet ist, die Einwanderung eosinophiler Zellen und die Umprogrammierung von Gewebemakrophagen in der Lunge von einem regenerativen Phänotyp zu einem proinflammatorischen Phänotyp, der mit einem Anstieg von IL-1, IL-6, IL-8, CXCL-10 und MCP-1 einhergeht.
IL-6, IL-8 da war doch was? IL-6 und IL-8 als Marker bei Post-Vac?
EN: Irrespectively of the putative pathogenetic role of receptor antibodies in PACVS, a combination of two index receptor antibodies (AT1R and α2b-adr-R) in conjunction with IL-6 allows discrimination of PACVS from the normal post-vaccination state with a cumulative sensitivity and specificity of up to 90%. However, increases in IL-6, IL-8 and AT1R antibodies have also been observed in long COVID-19 and post-COVID-19 ME/CFS.
DE: Unabhängig von der vermuteten pathogenetischen Rolle der Rezeptor-Antikörper bei PACVS ermöglicht eine Kombination von zwei Index-Rezeptor-Antikörpern (AT1R und α2b-adr-R) in Verbindung mit IL-6 die Unterscheidung von PACVS vom normalen Zustand nach der Impfung mit einer kumulativen Sensitivität und Spezifität von bis zu 90 %. Erhöhungen von IL-6-, IL-8- und AT1R-Antikörpern wurden jedoch auch bei langem COVID-19- und Post-COVID-19-ME/CFS beobachtet. (https://www.preprints.org/manuscript/202309.0113/v1).
Ich erinnere hier noch einmal an den oben erwähnten Wissensstand: 2007 stellte man bereits fest, dass das Spike-Protein stark inflammatorisch wirkt und zur Erhöhung der Entzündungsmarker IL-6 and TNF-alpha führt (https://pubmed.ncbi.nlm.nih.gov/17532082/),
Auf diese beiden Interleukine (IL) hat man doch angeblich laut PEI getestet bzw. hätte man unbedingt testen müssen. Wie auch immer, weil es gibt dafür kein Tiermodell, wie das PEI ja auch im nächsten zitierte Satz zugibt. Das hätte einem doch auffallen müssen, zumal ein Teil dieser Marker jetzt als Marker zur Diagnose von Impfschäden eingesetzt werden.
EN: The recording of these parameters is of central importance, since suitable animal models (non-human primates (NHP), ferrets, golden hamsters, and transgenic mice) for the reliable investigation and assessment of a potential ADE/ERD risk are currently still under development and are not yet fully qualified for the use as regulation-relevant methods.
DE: Die Erfassung dieser Parameter ist von zentraler Bedeutung, da geeignete Tiermodelle (nicht-menschliche Primaten (NHP), Frettchen, Goldhamster und transgene Mäuse) zur zuverlässigen Untersuchung und Bewertung eines potenziellen ADE/ERD-Risikos derzeit noch in der Entwicklung sind und sich noch nicht vollständig für den Einsatz als regulierungsrelevante Methoden eignen.
OK, nur mal zur Rekapitulation der Gedankenakrobatik, die dieser Satz dem PEI abverlangt haben muss. Man hat zuvor darauf bestanden geeignete Tiermodelle zu verwenden, um hier zuzugeben, dass es für eines der Hauptrisiken, die bei vorherigen Bemühungen aufgefallen waren (ADE), gar keine Tiermodelle gibt!
Die Marker, die vom PEI vorgeschlagen wurden, die man VORHER hätte prüfen müssen, werden JETZT zur Diagnose der Schäden eingesetzt.
Und wo wir noch einmal bei nicht vorhandenen Tiermodellen sind, für Myokarditis gibt es auch kein Tiermodell:

4. Anforderungen an klinische Prüfungen
EN: The dosage that proves to be optimal in terms of tolerability and immunogenicity is checked in the phase 2 clinical trial in a larger study group of several hundred healthy adults.
DE: Die Dosierung, die sich in Bezug auf Verträglichkeit und Immunogenität als optimal erweist, wird in der klinischen Studie der Phase 2 an einer größeren Studiengruppe von mehreren hundert gesunden Erwachsenen überprüft.
Theoretisch ja, praktisch nein. 20µg haben sich als optimal erwiesen, eingesetzt wurden 30µg.

Daran gibt es nicht zu diskutieren. Der Satz im PEI-Paper ist einfach nur falsch/gelogen.
EN: In addition, it is examined whether and to what extent the neutralizing antibody response decreases over time, since this is considered to be essential for the efficient and lasting protection against infection or disease.
DE: Darüber hinaus wird untersucht, ob und inwieweit die neutralisierende Antikörperreaktion im Laufe der Zeit abnimmt, da dies als wesentliche Voraussetzung für einen wirksamen und dauerhaften Schutz vor Infektionen oder Krankheiten angesehen wird.
Ja, das wurde überprüft und das schnelle Absinken dann einfach ignoriert:

https://twitter.com/StatChrisCotton/status/1639333283006644227
Auch die Preprints, die sich diesem Thema widmeten, hat das PEI einfach ignoriert: Large-scale study of antibody titer decay following BNT162b2 mRNA vaccine or SARS-CoV-2 infection | medRxiv
EN: The optimal study design for proof of effectiveness is currently being discussed internationally.
DE: Das optimale Studiendesign für den Nachweis der Wirksamkeit wird derzeit international diskutiert.
Dieser Satz wurde publiziert, nachdem die Menschen mit dem Werbespruch „sicher und effektiv“ in die Nadel getrieben wurden. Das Produkt war bereits seit Monaten auf dem Markt und in den Menschen und man diskutierte das optimale Studiendesign, um die Wirksamkeit nachzuweisen.
In diesem Zusammenhang verweise ich auf die oben diskutierten Daten der Cleveland Studie: Es gab keine Effektivität.
EN: Pursuant to this ordinance, the sponsor has to document in detail all adverse events reported to him by the investigators. These records should be sent to the Paul-Ehrlich-Institut upon request in pseudonymized form.
In addition, the sponsor has to inform the responsible ethics committee and the Paul-Ehrlich-Institut about every suspected case of an unexpected serious side effect (“serious unexpected suspected adverse reaction”, SUSAR), immediately and no later than 7 or 15 days after becoming known. This applies to SUSARs from all clinical trials with vaccines under investigation and also to clinical trials outside of Germany, provided that the same vaccine is being tested. The SUSAR reports are recorded in a database at the Paul-Ehrlich-Institut and assessed by a doctor with regard to causality. The obligation to report SUSARs also applies after the clinical trial has ended.
DE: Nach dieser Verordnung muss der Sponsor alle unerwünschten Ereignisse, die ihm von den Prüfärzten gemeldet werden, detailliert dokumentieren. Diese Unterlagen sind dem Paul-Ehrlich-Institut auf Anforderung in pseudonymisierter Form zuzusenden.
Darüber hinaus muss der Sponsor jeden Verdachtsfall einer unerwarteten schwerwiegenden Nebenwirkung ("serious unexpected suspected adverse reaction", SUSAR) unverzüglich, spätestens jedoch 7 bzw. 15 Tage nach Bekanntwerden, der zuständigen Ethik-Kommission und dem Paul-Ehrlich-Institut mitteilen. Dies gilt für SUSARs aus allen klinischen Prüfungen mit zu prüfenden Impfstoffen und auch für klinische Prüfungen außerhalb Deutschlands, sofern der gleiche Impfstoff geprüft wird. Die SUSAR-Meldungen werden im Paul-Ehrlich-Institut in einer Datenbank erfasst und von einem Arzt hinsichtlich der Kausalität bewertet. Die Meldepflicht für SUSARs gilt auch nach Beendigung der klinischen Prüfung.
Von dieser Datenbank habe ich noch nie gehört. Die Daten des PEI hätte ich gerne. Aber im englischen Original steht „should“. Entweder das ist wirklich das höfliche British English und drückt eine Pflicht aus, oder es ist die deutsche Variante von „sollte“ und es wird nicht eingefordert. Ich habe in all den Jahren noch nie von SUSAR Daten gehört. Die hätte ich aber gerne.
Da ich bei SUSARS nicht firm bin, hat mir hier dankensweiter Weise Hans-Joachim Kremer zugearbeitet:
In Sachen SUSAR wirkt der Text wie wenn im Fußball im eigenen Strafraum „schön“ gespielt wird: Allzu oft wird so vom Gegner bestraft, nicht folgt: Elfmeter – Tor.
Immerhin 328 Wörter verbraucht as PEI zu SUSARs, also suspected unexpected serious adverse reactions bzw. vermutete unerwartete schwerwiegende unerwünschte Reaktionen; trotz der Stellung am Anfang bezieht sich das suspected ausschließlich auf reactions. Dieses Konzept wurde Mitte der 1995 mit der Richtlinie ICH E2a international eingeführt, ist also schon lange etabliert. Aus den Regeln, die vom PEI - fast - korrekt wiedergegeben werden, ergeben sich praktisch keine Aufgaben für eine Behörde, aber viele für den Sponsor, hier also Pfizer-Biontech, Moderna usw. Für wen schrieb das PEI solche Banalitäten auf?
In Sachen Comirnaty kommt hinzu, dass in deren klinischer Studie kein einziger SUSAR auftrat, und zwar, weil kein einziger der 126 SAEs (events, Ereignisse) unter Comirnaty vom Prüfarzt in wenigstens einem möglichen Kausalzusammenhang (=reaction) mit der Impfung bewertet wurde. Noch seltsamer: 328 Wörter, obwohl absolut kein Arbeitsaufwand anfiel? Oder vielleicht doch, nur anders als wir denken?
Meine obige Einschränkung „fast“ hatte ja auch einen Sinn. Der bezog sich nämlich auf diese Aussage des PEI:
The SUSAR reports are recorded in a database at the Paul-Ehrlich-Institut and assessed by a doctor with regard to causality. (Die SUSAR-Meldungen werden im Paul-Ehrlich-Institut in einer Datenbank erfasst und von einem Arzt hinsichtlich der Kausalität bewertet.)
Der erste Teil ist in Ordnung, der zweite Teil des Satzes beschreibt aber eine Aufgabe, die einer Behörde gar nicht zusteht – oder besser gesagt, die sie sich hier anmaßt. Die Beurteilung der Kausalität obliegt nämlich dem Prüfarzt, und kann vom Sponsor eigentlich nur in Richtung höherer Kausalität verändert werden. Ein Eingriff und erst Recht eine Abwertung der Kausalität durch Behörden ist in den Regularien gar nicht vorgesehen. So heißt es in ICH E2a (1995):
EN: All cases judged by either the reporting health care professional or the sponsor as having a reasonable suspected causal relationship to the medicinal product qualify as ADRs.
DE: Als UAW gelten alle Fälle, bei denen entweder der meldende Angehörige der Gesundheitsberufe oder der Sponsor einen begründeten Verdacht auf einen ursächlichen Zusammenhang mit dem Arzneimittel sieht.
Allein schon wegen “all” gefolgt von „either … or“ gibt es keine Aufgabe der Behörden in Sachen Kausalitätsbewertung. Nicht nur, dass das PEI nach eigener Aussage, aber entgegen den Regularien, hier überhaupt eingriff ist problematisch. Erst recht problematisch ist diese Aussage, weil es im Ergebnis keinen einzigen SAR, also auch kein SUSAR unter den 126 SAEs gab, weist auf einen höchst bedenklichen Vorgang hin. Hat das PEI also aktiv dazu beigetragen, schwerwiegende Reaktionen in der randomisierten klinischen Studie zum Comirnaty unter den Teppich zu kehren?
EN: In addition to evaluating each individual report, the Paul-Ehrlich-Institut regularly conducts weekly statistical evaluations of the cumulative reports.
The sponsor will inform the Paul-Ehrlich-Institut and the responsible ethics committee immediately and no later than 15 days after becoming aware of any issues that require a renewed review of the risk–benefit assessment of the investigated medicinal product. For example, these include:
1. individual reports of expected serious adverse side effects with an unexpected outcome,
2. a clinically relevant increase in the frequency of expected serious adverse side effects,
3. Events connected to the conduct of the study or the development of the investigated medicinal product that could potentially affect the safety of the persons concerned.
The Paul-Ehrlich-Institut can request a list of all suspected serious side effects that occurred during the test as well as a report on the safety of the test participants, which otherwise has to be submitted annually.
DE: Neben der Auswertung jedes einzelnen Berichts führt das Paul-Ehrlich-Institut regelmäßig wöchentliche statistische Auswertungen der kumulierten Berichte durch.
Der Sponsor informiert das Paul-Ehrlich-Institut und die zuständige Ethik-Kommission unverzüglich, spätestens jedoch 15 Tage nach Bekanntwerden von Sachverhalten, die eine erneute Überprüfung der Nutzen-Risiko-Bewertung des untersuchten Arzneimittels erfordern. Dazu gehören beispielsweise:
1. Einzelberichte über erwartete schwerwiegende unerwünschte Nebenwirkungen mit unerwartetem Ausgang,
2. eine klinisch relevante Zunahme der Häufigkeit erwarteter schwerwiegender unerwünschter Nebenwirkungen,
3. Ereignisse im Zusammenhang mit der Durchführung der Studie oder der Entwicklung des untersuchten Arzneimittels, die möglicherweise die Sicherheit der betroffenen Personen beeinträchtigen könnten.
Das Paul-Ehrlich-Institut kann eine Auflistung aller vermuteten schwerwiegenden Nebenwirkungen, die während der Prüfung aufgetreten sind, sowie einen Bericht über die Sicherheit der Versuchsteilnehmer verlangen, der ansonsten jährlich vorzulegen ist.
Von diesen Daten habe ich auch noch nie gehört. Ich habe auch keinen einzigen dieser Berichte jemals gesehen. Es gab die RKI Updates, aber vom PEI gab es nichts, was mir bekannt wäre. Da sollte man mal nachhaken.
Sind damit vielleicht die rausgefragten PSURS gemeint, die man sich von der EU erstreiten musste, oder sind das ganz andere Datensätze?
EN: […], a sufficient database will be available to adequately assess quality, effectiveness, and safety. Nevertheless, after approval, further studies on the effectiveness (“effectiveness in everyday use”) and safety of the vaccines will also be carried out by the federal authorities in order to ensure that the vaccines retain their positive benefit–risk profile after approval in their intended usage in broad sections of the population.
DE: eine ausreichende Datenbasis zur Verfügung stehen wird, um Qualität, Wirksamkeit und Sicherheit angemessen zu bewerten. Dennoch werden auch nach der Zulassung weitere Studien zur Wirksamkeit ("Wirksamkeit im Alltag") und Sicherheit der Impfstoffe von den Bundesbehörden durchgeführt, um sicherzustellen, dass die Impfstoffe ihr positives Nutzen-Risiko-Profil auch nach der Zulassung in der vorgesehenen Anwendung in breiten Bevölkerungsschichten behalten.
Die Bewertung des PEI fiel irgendwie immer zugunsten der COVID-Produkte aus. Vielleicht wegen der falschen statistischen Methodik?

Mit „Observed versus Expected“ (https://www.pei.de/SharedDocs/Bilder/DE/newsroom/dossier/sicherheitsberichte/17-09-2021-tab-10.html) wird man nie ein Signal finden, habe ich mir von befreundeten Statistikern erklären lassen. Das ist aber nicht mein Metier und das hat mit Zahlen zu tun, daher kann ich nicht erklären, warum das so ist. Diese Vorgehensweise wurde jedenfalls als unseriös bezeichnet.
5. Marktzulassung und Verwaltungsangelegenheiten
Dieser fünfte Teil des Papers ist reines Blabla. Leere Worthülsen, die gut klingen und beruhigen sollen aber Inhaltsleer sind. Sie täuschen Sicherheit vor mit schönen Worten, die bei genauer Analyse nicht aussagen, nichts versprechen und in etwas so hart und verbindlich sind, wie Götterspeise, die man an die Wand zu nageln versucht.
EN: The assessment reports can be reviewed and commented on by all national authorities and, after discussion in the Committee for Human Medicinal Products (CHMP) of the EMA, are consolidated in one report. Open points are compiled in question lists and have to be addressed by the applicant in three question-and-answer rounds. The given period for the assessment and discussion by the authorities is up to a maximum of 210 days. The time the applicant needs to answer the questions is not counted towards this time.
DE: Die Beurteilungsberichte können von allen nationalen Behörden geprüft und kommentiert werden und werden nach Diskussion im Ausschuss für Humanarzneimittel (CHMP) der EMA in einem Bericht zusammengefasst. Offene Punkte werden in Fragelisten zusammengestellt und müssen vom Antragsteller in drei Frage-Antwort-Runden beantwortet werden. Die Frist für die Bewertung und Erörterung durch die Behörden beträgt maximal 210 Tage. Die Zeit, die der Antragsteller zur Beantwortung der Fragen benötigt, wird nicht auf diese Zeit angerechnet.
Die entsprechenden Daten sieht man wohl im EMA-Leak (https://t.co/fFsopN1R00) und wie das gelaufen ist. Wenn nach 3 Fragerunden nichts rauskam, wird die Akte geschlossen und der Antragsteller kann machen, wie er lustig ist. So scheint es mir, nach der Frage und Antwort Runde bezüglich des DNase I-Verdaus, den ich oben im Text bereits behandelt habe.
Und streng genommen steht hier im Text nichts anderes. Der Antragsteller hat 210 Tage zum antworten. Über Konsequenzen, die gezogen werden könnten, steht da nichts.
EN: The prerequisite is an unmet medical need in the case of serious illnesses, which can likely be covered by the new medicinal product. Therefore, accelerated evaluation is reserved for medicinal products that have clear advantages over already approved alternatives or are intended for indications for which no medicinal products are available.
DE: Voraussetzung ist ein ungedeckter medizinischer Bedarf bei schweren Krankheiten, der voraussichtlich durch das neue Arzneimittel gedeckt werden kann. Die beschleunigte Beurteilung ist daher Arzneimitteln vorbehalten, die eindeutige Vorteile gegenüber bereits zugelassenen Alternativen aufweisen oder für Indikationen bestimmt sind, für die es keine Arzneimittel gibt.
Die Stichworte hier wären Hydroxichloroquine (https://c19hcq.org/) und Ivermectin (https://c19ivm.org/desorthenin.html). Es gab immer erprobte, sichere, billige Alternativen. Die hat man aber unzugänglich gemacht, um diese neuen, experimentellen Produkte in den Markt zu drücken. Das ist aber eine andere Geschichte. Das aufzudröseln würde selbst ein ganzes Buch füllen.
EN: Even if the data are still incomplete, a risk–benefit analysis must still be possible.This “conditional marketing authorization” requires that the existing gaps in the database are closed after approval. […] In most cases, this refers to conducting or completing clinical trials.[…] In the present case of the SARS-CoV-2 pandemic, a medicinal product could therefore also be approved if the pharmaceutical development has not yet been completed in all details. Here, too, the authorization holder must close these gaps in the database after authorization. This “conditional admission” is valid for one year and can be extended several times.
DE: Auch wenn die Daten noch unvollständig sind, muss eine Nutzen-Risiko-Analyse möglich sein. Diese "bedingte Zulassung" setzt voraus, dass die bestehenden Lücken in der Datenbasis nach der Zulassung geschlossen werden. […] In den meisten Fällen handelt es sich dabei um die Durchführung oder den Abschluss von klinischen Studien. […] Im aktuellen Fall der SARS-CoV-2-Pandemie könnte also ein Arzneimittel auch dann zugelassen werden, wenn die pharmazeutische Entwicklung noch nicht in allen Details abgeschlossen ist. Auch hier muss der Zulassungsinhaber diese Lücken in der Datenbank nach der Zulassung schließen. Diese "bedingte Zulassung" ist für ein Jahr gültig und kann mehrfach verlängert werden.
Das sah dann so aus in der Realität (https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf S. 140)

3 Jahre nach Markteinführung sollten dann die Daten der klinischen Studie vorliegen, während in der Zeit bereits geboostert wurde und “mix and match” betrieben wurde, ganz ohne Datenlage.
Das Ergebnis war auch entsprechend unschön, als unabhängige Wissenschaftler sich die Daten der Studie vornahmen:
https://drbine.substack.com/p/der-heie-schei-teil-4-todesmeldungen

https://twitter.com/DrJKunadhasan/status/1714888450116386929 und https://doi.org/10.56098/ijvtpr.v3i1.85
Nur Konsequenzen hatten diese Daten bisher leider keine.
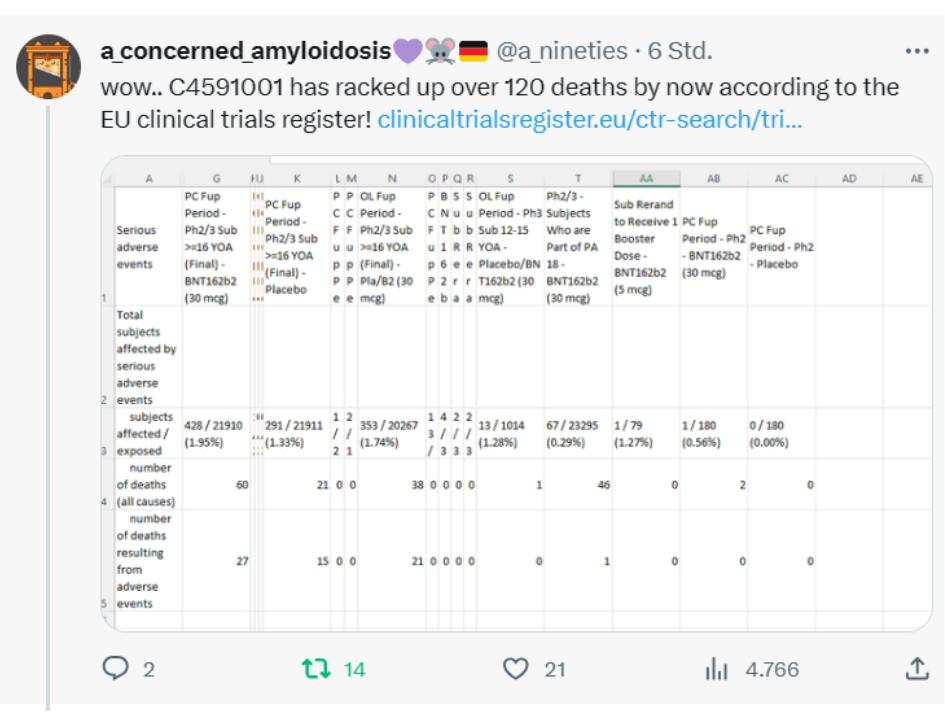
https://twitter.com/a_nineties/status/1708785215106134257/photo/1
https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-002641-42/results#adverseEventsSection
EN: Long-term monitoring of the vaccine’s immunogenicity is also an obvious requirement.
DE: Auch die langfristige Überwachung der Immunogenität des Impfstoffs ist eine offensichtliche Notwendigkeit.
Damit sind wohl die PSURS gemeint. Deren katastrophale Sicherheitsdaten hatten aber leider nie irgendwelche Konsequenzen, für keine der Firmen.
Immunogenität ist übrigens nicht Immunität. Immunogenität heißt nur, dass eine Immunreaktion ausgelöst wird (https://flexikon.doccheck.com/de/Immunogenit%C3%A4t). Welcher Art diese ist, das kann sehr unterschiedlich sein. Darunter müsste eigentlich auch der Shift zu IgG4 Antikörpern fallen, die zur Toleranz gegen das Spike-Protein führen.
Aber erneut, langfristige Überwachung ohne angedrohte Konsequenzen. Überwacht wird es wohl. Nur eben ohne Folgen für irgendwen, weder für die Firmen, noch das PEI noch die Patienten, die das Produkt erhalten haben. Das ist wie Überwachungskameras, deren Videos nie angesehen werden. Nutzlos. Leere Worte. Schaumschlägerei. Politisches Schattenboxen.
EN: With this approval variant, it is also possible to impose conditions that serve to re-evaluate the benefit–Risk profile. As with the conditional approval, there is an annual reporting obligation of the approval holder.
DE: Auch bei dieser Zulassungsvariante ist es möglich, Auflagen zu erteilen, die der Neubewertung des Nutzen-Risiko-Profils dienen. Wie bei der bedingten Zulassung besteht eine jährliche Berichtspflicht des Zulassungsinhabers.
Windelweichere Formulierungen gehen kaum noch.
„Ist es möglich“ = kann man machen, muss man aber nicht.
„Auch bei dieser Zulassungsvariante ist es möglich, Auflagen zu erteilen, die der Neubewertung des Nutzen-Risiko-Profils dienen“ = Hier steht nichts, dass die Auflagen erfüllt werden müssen. Keine Konsequenzen bei Nichterfüllung. Auch die Neubewertung des Nutzen-Risiko-Profils zieht keinerlei Konsequenzen nach sich. Es werden keine Parameter genannt, welche Konsequenzen oder Strafen beinhalten würden. Man erteilt halt Auflagen, die erfüllt werden oder eben nicht, um dann eine Riskikoanalyse durchzuführen, die keinerlei Konsequenzen für niemanden hat.
Der größte Witz ist der Disclaimer am Schluss. Die Autoren haben keine Interessenskonflikte.

Ich verweise an dieser Stelle auf Teil 1 dieses Artikels:
https://drbine.substack.com/p/wie-man-sich-aus-seinen-eigenen-worten-a18
Keine Interessenskonflikte sieht, meiner Meinung nach, anders aus.
Immerhin, alle Autoren haben ihre Autorenschaft gestanden und dass sie eingewilligt haben, dass dieses Lügenkonstrukt veröffentlicht wird. Hier kann sich keiner rauswinden.
Updates:
Wie ist es zu erklären dass all diese Leute und Institutionen - PEI, EMA, Wissenschaftsjournale und ihre Herausgeber und Redaktionen mitgemacht haben ?
https://horizons.service.canada.ca/en/2021/07/29/what-is-the-biodigital-convergence/index.shtml
Via Corbetts doku:
Hier ist alles drin zum Thema Transhumanismus und synthetische Biologie, bis hin zu Links zum Implantationsrobotor von Neuralink aus der Werkstatt von Lawrence Livermore wo bekanntlich auch Atomwaffen herkommen. War ja neulich in der Radiowerbung hier. Dazu brauche ich keine esoterischen oder religiösen Konzepte. Irgendwann Standard für Piloten und wer weiss wer noch.