

Disclaimer:
Es mag nach diesem Artikel vielleicht so klingen, dass die Mitochondriopathie die Ursache für alle Nebenwirkungen sein könnte. ABER VORSICHT! Der Körper hat N Möglichkeiten sich zu äußern. Diesen Symptomen können aber X verschiedene Ursachen zugrunde liegen.
Viele Autoren erliegen leider der Versuchung zu glauben, sie hätten die Ursache gefunden, die man behandeln muss und das Problem ist gelöst. Es gibt aber zu viele Mechanismen da draußen, die alle jeweils alleine die Symptome erklären könnten. Beispiele wären cGAS/STING, Ras-Kaskade, Endotoxine… ABER, wenn diese Mechanismen parallel/gleichzeitig aktiv sind, wird es immer noch nur die N möglichen Symptome geben, denen X verschiedene Schäden zugrunde liegen.
Daher, lieber Leser, die Mitochondriopathie ist nur ein weiterer Mechanismus von vielen, die ausgelöst werden und nicht die alleinige Ursache, die es zu behandeln gilt, nur eine der Ursachen, eine der vielen Ursachen, die alle bekannten Symptomatiken bereits alleine erzeugen kann.
Als Grundgerüst für diesen Artikel nehme ich das Review von J. Finsterer.
Finsterer J. Mitochondriopathies. Eur J Neurol. 2004 Mar;11(3):163-86. doi: 10.1046/j.1351-5101.2003.00728.x. PMID: 15009163. https://pubmed.ncbi.nlm.nih.gov/15009163/
1:1 Übersetzungen und gekürzte Übersetzungen aus diesem Paper sind in kursiv, aber ohne jeweils das Zitat dahinter zu schreiben. Ich bauen diesen Substack um das Finsterer Review herum auf und ergänze mit Lehrbuchdaten, wo Grundlagen gebraucht werden und neueren Daten, da das Finsterer Review schon ein wenig älter ist.
Mitochondrien Grundlagen
Aufbau der Mitochondrien
Mitochondrien sind intrazelluläre kugelförmige oder eiförmige Organellen mit einem Querdurchmesser von 0,1-0,5 µm und einer variablen Länge. Nach der Endosymbiontenhypothese stammen die Mitochondrien von Prokaryoten ab, die in kernhaltige Zellen integriert wurden. Die Anzahl der Mitochondrien pro Zelle reicht von keinem (Erythrozyten) bis zu 10 000 (quergestreifte Muskelzelle). Die durchschnittliche Anzahl liegt bei 500-2000/Zelle. Die Anzahl der Mitochondrien in einer Zelle steigt mit der Menge an Substrat und Sauerstoff, die eine bestimmte Zelle benötigt. In der Muskelzelle befinden sich die Mitochondrien zwischen den Myofibrillen, subsarcolemmal, in der Nähe des Zellkerns oder in der Nähe der motorischen Endplatte. Mitochondrien sind aus vier Kompartimenten aufgebaut:
(i) der äußeren Membran,
(ii) der inneren Membran,
(iii) dem Intermembranraum
(iv) die Matrix

(Campbell Biology, 12th Edition, Ausschnitt aus Abbildung 6.17)
Aufgaben der Mitochondrien
Mitochondrien erfüllen vier grundlegende biologische Aufgaben:
1. Bereitstellung von ATP
2. Vermittlung des Zelltods durch Apoptose
3. Wärmeproduktion
4. Beitrag zur Humangenetik. Darüber hinaus beherbergen Mitochondrien die β-Oxidation, den Citrat-Zyklus (Tricarbox-Zyklus, Krebs-Zyklus), den Abbau von Aminosäuren, Teile der Häm-Biosynthese, Teile des Steroid-Stoffwechsels, Teile des Harnsäure-Zyklus, die mitochondriale Proteinsynthese und den Pyruvat-Dehydrogenase-Komplex (PDC), der für die Decarboxylierung von Pyruvat verantwortlich ist.
Bereitstellung von ATP:
Im Mitochondrium laufen mehrere Energiegewinnungsoptionen parallel, wie das Schaubild aus dem alten Campbell Biologie, Spektrum Verlag, 1. Auflage von 1998 schön übersichtlich zeigt.
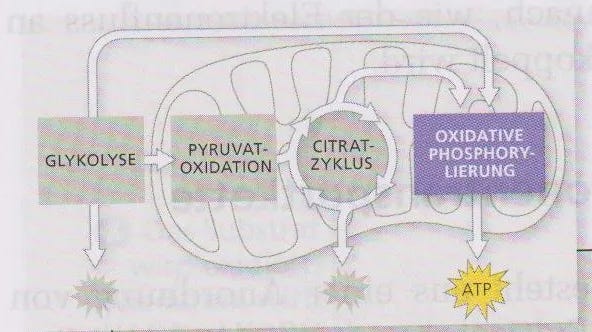
In der sogenannten Atmungskette werden Elektronen durchgereicht und auf immer niedrigere Eerngieniveaus abgesenkt und die freiwerdende Energie an ATP gekoppelt, die Energiewährung des Körpers. Man könnte die Atmungskette auch eine kontrollierte Knallgasreaktion nennen.
Mitochondrien und die Chloroplasten aus den Pflanzen sind sich dabei sehr ähnlich, wobei bei den Chloroplasten die Energie aus der Sonne kommt.

Die Energieänderungen der Elektronen während ihres linearen Flusses durch die Lichtreaktionen sind in Abbildung 10.14 in einer mechanischen Analogie dargestellt. Obwohl das Schema in Abbildungen 10.13 und 10.14 dargestellte Schema kompliziert erscheinen mag, sollten Sie das große Ganze nicht aus den Augen verlieren: Die Lichtreaktionen nutzen die Sonnenenergie, um ATP und NADPH zu erzeugen, die chemische Energie bzw. Reduktionskraft für die kohlenhydratsynthetisierenden Reaktionen des Calvin-Zyklus liefern. (Campbell Biology, 12th Edition)

Vermittlung des Zelltods durch Apoptose
Cytochrom C, initiiert die Apoptose, wenn es in das Zytosol der Zelle freigesetzt wird. Das ist natürlich komplexer und beinhaltet diverse Moleküle und Signalketten, die aber ohne Cytochrom C nicht gestartet werden, es sei denn von Extern. Cytochrom C ist für den intrinsischen Selbstmord zuständig. Fällt Cytochrom C aus muss die Zelle von außen getötet werden bzw. geht in einen Entzündungszustand mit reaktiven Sauerstoffspezies (ROS) über, was neben Entzündungen auch Thrombosen nach sich ziehen kann.

Abb. 1 | Apoptotische Signalwege. Die Apoptose kann über zwei Wege erfolgen: extrinsisch und intrinsisch. Der extrinsische (auch als Todesrezeptor bezeichnete) apoptotische Weg beinhaltet die Bindung eines Todesrezeptor-Liganden an ein Mitglied der Todesrezeptorfamilie (Mitglieder der Tumornekrose-Rezeptor-Superfamilie). Die Bindung des Fas-Liganden (FAS-L) an FAS beispielsweise löst die Apoptose aus, indem das Adaptormolekül Fas-associated death domain (FADD) rekrutiert wird. FADD bindet an den Initiator Caspase 8 und induziert dessen Dimerisierung, was zu seiner Aktivierung führt. Die aktive Caspase 8 spaltet und aktiviert die Ausführ-Caspasen 3 und 7, was zu einer weitreichenden Spaltung von Zellbestandteilen und einem schnellen Zelltod führt. Der intrinsische (auch als mitochondrialer) apoptotische Weg wird durch eine Vielzahl unterschiedlicher Stimuli ausgelöst (u. a. DNA-Schäden, Wachstumsfaktorentzug und mitotischer Stillstand), die eine Aktivierung von BH3-only-Mitgliedern der B-Zell-Lymphom 2 (BCL-2)-Proteinfamilie bewirken. Nur-BH3-Proteine hemmen anti-apoptotische BCL-2-Proteine und aktivieren die pro-apoptotischen BCL-2-Effektorproteine BAX und BAK, was zu einer Permeabilisierung der mitochondrialen Außenmembran (MOMP) führt. Dies ermöglicht die Freisetzung von Proteinen aus dem mitochondrialen Intermembranraum, die Caspasen aktivieren, insbesondere Cytochrom C. Cytochrom C bindet an den apoptotischen Peptidase-Aktivierungsfaktor 1 (APAF1) und bildet eine eptamere Struktur, das sogenannte Apoptosom. Dieses rekrutiert und aktiviert den Initiator Caspase 9, der Caspase 3 und Caspase 7 spaltet und aktiviert. MOMP bewirkt auch die Freisetzung von Proteinen wie SMAC und OMI, die den Caspase-Inhibitor XIAP blockieren und so die Apoptose fördern. Die durch Caspase 8 vermittelte Spaltung und Aktivierung des BH3-only-Proteins BID (zur Erzeugung von tBID) verbindet den extrinsischen apoptotischen Weg mit dem intrinsischen Weg. (https://pubmed.ncbi.nlm.nih.gov/31636403/)
Gestresste Zellen müssen aber nicht sofort in den Selbstmord getrieben werden. Man kann sie auch sublethal stressen. Das führt dann z. Bsp. zu Entzündungen.

Abb. 3 | Unterschiedliche Grade der Permeabilisierung der mitochondrialen Außenmembran ermöglichen das Überleben der Zelle und demaskieren Signalfunktionen. Apoptotischer Stress kann zu einer unvollständigen Permeabilisierung der mitochondrialen Außenmembran (MOMP) führen, die mit dem Überleben der Zelle vereinbar ist. a | Zellen, die zur Apoptose veranlasst wurden, können unter Bedingungen der Caspase-Inhibition überleben. Das Überleben der Zelle in diesem Zusammenhang erfordert das Vorhandensein einer Subpopulation intakter Mitochondrien, die keine MOMP durchlaufen haben. Das Überleben der Zelle hängt auch von der Expression der Glyceraldehyd-3-Phosphat-Dehydrogenase (GAPDH) ab, die eine hohe glykolytische Aktivität und Autophagie unterstützt und dadurch Energie erzeugt, um eine metabolische Katastrophe zu verhindern und dysfunktionale Mitochondrien zu entfernen, die weitere Schäden verursachen könnten. Durch diese Mechanismen können die Zellen lange genug überleben, um den intakten Mitochondrien die Möglichkeit zu geben, sich zu vermehren und so das Überleben der Zelle zu ermöglichen. b | Subletale Belastungen, z. B. eine BH3-mimetische Behandlung, können dazu führen, dass nur eine Teilmenge der Mitochondrien MOMP durchläuft - ein Zustand, der als Minority MOMP bekannt ist. Minority MOMP kann eine begrenzte, subletale Caspase-Aktivität auslösen, die mit DNA-Schäden verbunden ist, die von Caspase-aktivierter DNase (CAD) und Caspase-3-abhängiger Freisetzung von Endonuklease G (endoG) aus Mitochondrien abhängen. Solche DNA-Schäden können eine onkogene Transformation fördern. Minoritäts-MOMP kann auch ohne Zelltod entzündungsfördernde Signale auslösen, z. B. durch Induktion CAD-abhängiger DNA-Schäden oder durch Freisetzung von mtDNA, die beide über die zyklische GMP-AMP-Synthase (cGAS) - Stimulator von Interferon-Genen (STING) entzündungsfördernde Signale auslösen können. (https://pubmed.ncbi.nlm.nih.gov/31636403/)
cGAS/STING ist also Teil des Problems, aber nicht die alleinige Lösung oder Ursache, denn Cytochrom-C und somit die Mitochondrien haben da auch einen Beitrag geleistet. Das meinte ich im Disclaimer mit dem Problem, dass es nicht eine Ursache und somit eine mögliche einfache Lösung gibt.
Und wir lernen auch, dass gestresste Mitochondrien, die nicht den Zelltod auslösen die Zelle unter umständen zu einer Krebszelle machen können oder zumindest Entzündungen auslösen. Prinzipiell wäre die Zelle aber noch zu retten, wenn es noch unbeschädigte Mitochondrien gibt. Die Biologie arbeitet also nicht nach einem entweder/oder-Prinzip sondern hält sich Hintertüren auf, die aber zu Folgeschäden führen können.
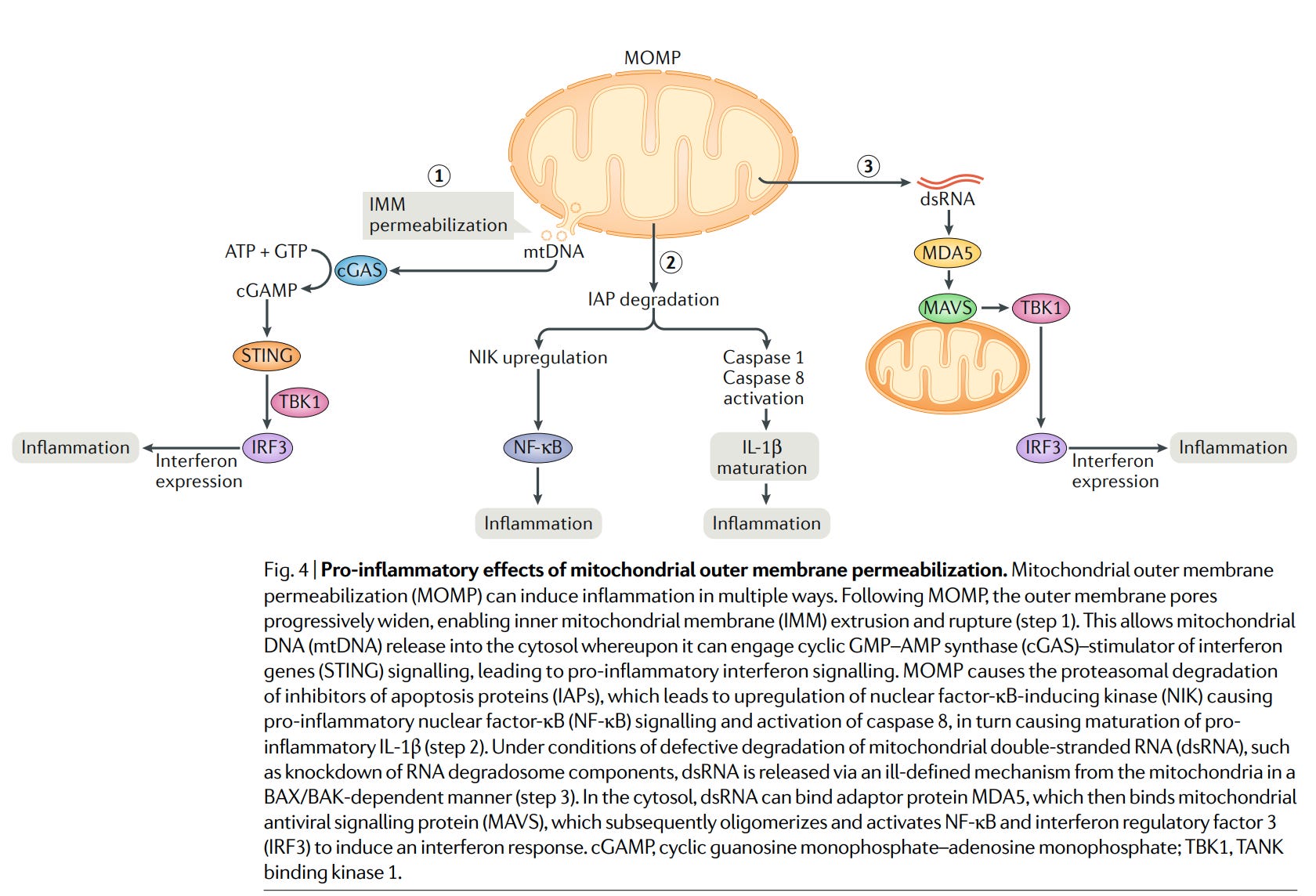
Abb. 4 | Entzündungsfördernde Effekte der Permeabilisierung der mitochondrialen Außenmembran. Die Permeabilisierung der mitochondrialen Außenmembran (MOMP) kann auf verschiedene Weise eine Entzündung auslösen. Nach der MOMP erweitern sich die Poren der äußeren Membran schrittweise erweitert, wodurch die innere mitochondriale Membran (IMM) extrusioniert und gerissen wird (Schritt 1). Dies ermöglicht die Freisetzung mitochondrialer DNA (mtDNA) in das Zytosol freigesetzt werden, woraufhin sie die zyklische GMP-AMP-Synthase (cGAS) - Stimulator der Interferon Genen (STING) in Gang setzen kann, was zu einer entzündungsfördernden Interferon-Signalisierung führt. MOMP bewirkt den proteasomalen Abbau von Apoptoseinhibitoren (IAPs), was zu einer Hochregulierung der Nuklearfaktor-κB-induzierenden Kinase (NIK) führt, die pro-inflammatorischen Nuklearfaktor-κB-Signalen (NF-κB) und der Aktivierung von Caspase 8, was wiederum die Reifung von pro-inflammatorischem IL-1β bewirkt (Schritt 2). Unter Bedingungen, unter denen der Abbau von mitochondrialer doppelsträngiger RNA (dsRNA) gestört ist, wie Knockdown von RNA-Degradosom-Komponenten, wird dsRNA über einen nicht genau definierten Mechanismus aus den Mitochondrien freigesetzt, und zwar BAX/BAK-abhängige Weise aus den Mitochondrien freigesetzt (Schritt 3). Im Zytosol kann dsRNA das Adaptorprotein MDA5 binden, das dann an das mitochondriale antivirales Signalprotein (MAVS) bindet, das anschließend oligomerisiert und NF-κB sowie den Interferon-Regulationsfaktor 3 (IRF3) aktiviert, um eine Interferonantwort auszulösen. cGAMP, zyklisches Guanosinmonophosphat-Adenosinmonophosphat; TBK1, TANK bindende Kinase 1
(https://pubmed.ncbi.nlm.nih.gov/31636403/)

Abb. 6|Mitochondrien und nicht-apoptotischer Zelltod.
a| Nekroptose ist eine entzündungsfördernde Form des Zelltods, die mit der Freisetzung von schadensassoziierten molekularen Mustern (DAMPs) einhergeht. Verschiedene Behandlungen können Nekroptose auslösen, die am besten nach einer Behandlung mit Tumornekrosefaktor (TNF) charakterisiert ist. Unter Hemmung der Caspase führt die TNF-Behandlung zu einer sequentiellen Phosphorylierung (P) und Aktivierung der rezeptorinteragierenden Proteinkinase 1 (RIPK1) und RIPK3 sowie zur Bildung von Nekrosomen. Das Nekrosom phosphoryliert und aktiviert dann die Pseudokinase Mixed-Lineage Kinase Domain-like Pseudokinase (MLKL), die zur Plasmamembran transloziert und diese permeabilisiert, wodurch die Zelle getötet wird. RIPK3 aktiviert auch den mitochondrialen Pyruvatdehydrogenase (PDH)-Komplex, was zu einer verstärkten aeroben Atmung und einer erhöhten Bildung reaktiver Sauerstoffspezies (ROS) führt. Diese aus den Mitochondrien stammenden ROS können den Aufbau von Nekrosomen und die Aktivität von RIPK3 verstärken.
b| Die Aktivierung von Inflammasom-Komplexen, z. B. durch intrazelluläre Krankheitserreger, führt zur Aktivierung von inflammatorischen Caspasen, die die proinflammatorischen Zytokine IL-1β und IL-18 spalten, was zu deren Reifung führt. Entzündliche Caspasen spalten und aktivieren auch Gasdermin D (GSDMD). Aktives GSDMD bildet Poren und permeabilisiert die Plasmamembran, was zum pyroptotischen Zelltod führt. Aktives GSDMD kann auch eine Permeabilisierung der äußeren Membran der Mitochondrien (MOMP) verursachen. Zusätzlich, Inflammasom-Aktivität die MOMP durch Spaltung und Aktivierung des BH3-only-Proteins BID. Stromabwärts von MOMP führt die Aktivierung von Caspase 3 zu einer spaltungsabhängigen Aktivierung des Kaliumkanal-bildenden Glykoproteins Pannexin 1. Dies bewirkt einen Kalium-Efflux aus der Zelle, was den Aufbau des Inflammasoms fördert.
c | Ferroptose wird durch oxidierte Lipide in Reaktionen ausgelöst, die mit Hilfe von Eisen und ROS katalysiert werden (Fenton-Reaktion). Der Schutz vor dieser Reaktion wird durch die Glutathionperoxidase 4 (GPX4) gewährleistet, die schädliche Lipidperoxide inaktiviert. Eine Möglichkeit zur Induktion der Ferroptose besteht in der Behandlung mit Erastin, das die Einfuhr von Cystein blockiert und die GPX4-Aktivität beeinträchtigt, oder in einem Cystein-Entzug. Neben der Beeinträchtigung von GPX4 führt der Cysteinmangel auch zu einer verstärkten Glutaminolyse, die den mitochondrialen Tricarbonsäurezyklus (TCA) speist, wodurch die mitochondriale Atmung erhöht wird und infolgedessen die mitochondriale ROS-Konzentration ansteigt. Eisen wird in verschiedenen eisenbindenden Proteinen gespeichert, darunter Ferritin und hämhaltige Proteine, und die Mitochondrien tragen zu dieser Speicherung bei. Diese eisenspeichernden Proteine werden unter bestimmten, den Zelltod auslösenden Bedingungen abgebaut, was zu einer Eisenfreisetzung führt. Die Nähe der Mitochondrienmembranen zu solchen Quellen von freiem Eisen und ROS macht sie zu einem wichtigen Ziel für die mit der Ferroptose verbundene Lipidoxidation. ETC, Elektronentransportkette; GSH, Glutathion. (https://pubmed.ncbi.nlm.nih.gov/31636403/)
Das Inflammasom kann übrigens auch Thrombosen auslösen. Damit könnte eine Entzündung der Zelle durch geschädigte Mitochondrien langfristig über das Inflammasom oder cGAS/STING zu Thrombosen führen.
X Mechanismen der Thrombosenbildung durch die Plörre

Mögliche Thrombosemechanismen durch die COVID-Injektionen Es ist mittlerweile hinlänglich bekannt, dass die COVID-Injektionen Thrombosen auslösen. Die Erkenntnis um die Vielfalt der Mechanismen jedoch, durch welche das geschehen kann, wächst von Monat zu Monat.
Das Spike Protein stört die Mitochondrien und führt zu mehr ROS:
„Die 24-stündige S1-Behandlung erhöhte die ATP-Produktion und die mitochondriale Atmung, indem sie die Expression von Fettsäure-transportierenden Regulatoren erhöhte und ein negativeres mitochondriales Membranpotential (Δψm) induzierte. Die 72-stündige S1-Behandlung verringerte die mitochondriale Atmungsrate und Δψm, erhöhte jedoch den Gehalt an reaktiven Sauerstoffspezies (ROS), mCa2+ und intrazellulärem Ca2+. Die Elektronenmikroskopie zeigte eine erhöhte mitochondriale Fragmentierung/Spaltung in AC16-Zellen, die 72 Stunden lang behandelt wurden.“ (https://pubmed.ncbi.nlm.nih.gov/36980218/)
Deregulierung der RAS führt zu ROS
ROS/Spike-Protein/Mitochondrien und wie sie die Alterung beschleunigen sind Walter M Chesnuts Spezialität, er geht da mehr ins Detail. Das würde hier den Rahmen sprengen.
Kurzversion:
Die Bindung des Spike Proteins an ACE2 führt zu einer Gleichgewichtsverschiebung im für die Regulation von Körperfunktionen extrem wichtigen und zentralen Renin-Angiotensin System (RAS), was zu eine Überschuss von Ang II führt, welches den gefäßverengenden Rezeptor vom Typ 1 (AT1R) aktiviert. (https://pubmed.ncbi.nlm.nih.gov/35408447/)

(https://pubmed.ncbi.nlm.nih.gov/37711109/)
Angiotensin II (Ang II) erhöht zudem die Produktion von reaktiven Sauerstoffspezies (reactive oxygene species ROS)-Produktion durch AT1R-abhängige Induktion von NADPH-Oxidase (Nox), was zu mitochondrialer Dysfunktion und DNA-Schäden führt und die Induktion von Entzündungszytokinen durch NF-κB unterstützt.

(https://pubmed.ncbi.nlm.nih.gov/16870827/)

(https://pubmed.ncbi.nlm.nih.gov/37711109/)
Ausführlicher habe ich das in diesem Artikel behandelt:
Deregulation von praktisch allem über RAS und AT1R

Disclaimer und Spoiler: Es mag nach diesem Artikel vielleicht so klingen, dass die RAS-Kaskade die Ursache für alle Nebenwirkungen sein könnte. ABER VORSICHT! Der Körper hat N Möglichkeiten sich zu äußern. Diesen Symptomen können aber X verschiedene Ursachen zugrunde liegen.
Autoimmunangriff auf die Mitochondrien.
Professor Gunther Hartmann von der Uni Bonn zeigt in einer seiner aktuelle Studie aus dem Februar 2024 mit dem Namen „Chronischer Stress des endoplasmatischen Retikulums bei myotoner Dystrophie Typ 2 fördert Autoimmunität durch Freisetzung mitochondrialer DNA“ (https://pubmed.ncbi.nlm.nih.gov/38378748/), dass Muskeln durch einen Autoimmunangriff zerstört werden, wenn das Endoplasmatische Retikulum gestresst wird und dadurch mitochondriale RNA (mtRNA) freigesetzt wird.
„Insgesamt zeigt unsere Studie einen neuen Mechanismus, durch den große Wiederholungsexpansionen chronischen endoplasmatischen Retikulumstress und damit verbundenen mtDNA-Austritt verursachen. Diese mtDNA wird wiederum durch den cGAS/STING-Signalweg erkannt und löst eine Typ-I-IFN-Reaktion aus, die zu Autoimmunität führt. Die Aufklärung dieses Weges eröffnet neue potenzielle therapeutische Ansatzpunkte für Autoimmunerkrankungen, die mit der Wiederholungsexpansion in Zusammenhang stehen.“ (https://pubmed.ncbi.nlm.nih.gov/38378748/)
1. Das Spike-Protein wird laut BioNTech eigenen Unterlagen im Endoplasmatischen Retikulum produziert (ER) (https://www.tga.gov.au/sites/default/files/foi-2389-03-1.pdf und tga.gov.au/sites/default/files/foi-2389-06.pdf)
2. Es konnte mittels Raman-Spektroskopie auf zellulärer Ebene gezeigt werden, dass das ER nach der modRNA-Impfung Schaden nimmt (https://www.biorxiv.org/content/10.1101/2022.03.02.482639v1.full.pdf). Dieses Ergebnis verwirrte die Autoren der Studie, da diese fest davon ausgingen, dass das Spike-Protein im Zytosol produziert wird, was falsch ist. Umso zuverlässiger ist deren somit kontraintuitive Messung, die der Hypothese der Autoren widerspricht. Mit dem Wissen, dass das Spike-Protein im ER produziert wird, ist die Messung schlüssig.
3. Der durch den Stress durch permanente Spike-Produktion in hoher Konzentration im ER verursachte Austritt von mtRNA führt zu einer Autoimmunreaktion wegen eines erworbenen ER-Stresses, der sich über Jahre hinziehen kann, da die Spike-Produktion bei vielen Menschen in diversen Organen immer noch aktiv ist und nicht stoppt.
4. Dieser Autoimmunangriff kann somit theoretisch in jedem Zelltyp passieren, der unter dauerhaftem ER-Stress leidet.
Es können sich aber auch einfach Antikörper gegen des Spike-Protein statt dessen gegen die Mitochondrien richten, weil das Spike-Protein aufgrund molekularer Mimikry Ähnlichkeit zu diversen menschlichen Proteinen aufweist.

(https://pubmed.ncbi.nlm.nih.gov/33584709/)
Die Vermutungen der zu humanen Proteinstrukturen ähnlichen Anteile des Spike Proteins variieren zwischen mehr als zwei Dutzend Hepta- und Oktamere (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8387358/) und bis zu 78.4% bei Fragmenten von 5-6 Aminosäuren (https://pubmed.ncbi.nlm.nih.gov/34192262/).
Die Hinweise auf molekulare Mimikry, welche bekannt dafür ist, zu Autoimmunangriffen zu führen (https://pubmed.ncbi.nlm.nih.gov/30509385/), verdichten sich immer mehr (https://doi.org/10.1101/2024.02.15.24302857, https://pubmed.ncbi.nlm.nih.gov/33584709/, https://www.mdpi.com/1999-4915/14/7/1415)
Das Spike Protein selbst schädigt die Mitochondrien

“Soweit wir wissen, ist dies die erste Studie, die die Auswirkungen von S1 auf die mitochondriale Funktion in menschlichen Kardiomyozyten untersucht. Obwohl S1 die mitochondriale Funktion in AC16-Zellen kurzfristig verbesserte, führte eine längere S1-Behandlung zu mitochondrialer Dysfunktion aufgrund der Unterbrechung von ∆ψm, mitochondrialer Ca2+-Überladung, Akkumulation von ROS und einer Veränderung der mitochondrialen Dynamik. Diese Auswirkungen der Untereinheit S1 des S-Proteins führen zu einem irreversiblen kardialen Remodeling.”
(https://pubmed.ncbi.nlm.nih.gov/36980218/)
Die Autoren stellen auch eine mechanistische Hypothese auf, wie das passieren könnte.

“Vorgeschlagene Mechanismen, durch die S1 eine mitochondriale Dysfunktion des Herzens auslöst, die zu Herzschäden bei COVID-19-Patienten führt.
Das Spike-Protein ist das glykosylierte Protein, das die Oberfläche von SARS-CoV-2 bedeckt und an den ACE2-Rezeptor des Wirts bindet, um den Eintritt des Virus in die Zelle zu vermitteln. Es besteht aus den Untereinheiten S1 und S2, die für die ACE2-Bindung bzw. die Membranfusion verantwortlich sind. S1 bindet möglicherweise an ACE2 auf der AC16-Membran und wird dann in das Zytosol internalisiert und in Organellen wie Mitochondrien lokalisiert, was den vorübergehenden Anstieg des Fettsäuretransports und der Aufnahme für die Biogenetik, Δψm und permanentes mCa2+ bewirkt und die Δψm später, was schließlich die mitochondriale Funktion beeinträchtigt und die ROS-Produktion fördert. ROS wiederum verschlechtern die mitochondriale Funktion und die mitochondriale Fragmentierung weiter. Darüber hinaus bewirkt S1 eine Herunterregulierung von TOM20; dieser Effekt könnte die Signalwege hemmen, die zur mitochondrialen Biogenese führen.
ACE2, Angiotensin-Converting-Enzym 2; FAT, Fettsäure-Translocase; PCT1/2, Carnitin-Palmitoyltransferase 1/2; MCD, Malonyl-CoA-Decarboxylase; ACC, Acetyl-CoA-Carboxylase; AMPK, AMP-aktivierte Proteinkinase; mCa2+, mitochondriales Kalzium, Δψm, mitochondriales Membranpotenzial; ROS, reaktive Sauerstoffspezies.“
Mitochondriale DNA
Mitochondrien haben ihr eigenes funktionelles Genom, das vom Kerngenom getrennt ist. Die MtDNA liegt innerhalb der mitochondrialen Matrix und weist strukturelle und funktionelle Merkmale auf, die sich von denen der nDNA unterscheiden (Tabelle 2). MtDNA ist ein zirkuläres, doppelsträngiges (schwerer (H, reich an Guaninen) und leichter (L, reich an Cytosinen) Strang) DNA-Molekül, das aus 16 569 Basenpaaren besteht. MtDNA-Gene kodieren für zwei rRNAs, 22 tRNAs und 13 Polypeptide (28 auf dem H-Strang und neun auf dem L-Strang). Im Gegensatz zur nDNA haben die kodierenden Sequenzen der mtDNA keine Introns (93 % sind kodierend, im Vergleich zu 5 % im Kerngenom), die mtDNA ist nicht mit Histonen verwoben, hat kein effektives Reparatursystem und verwendet einen abweichenden genetischen Code (Tabelle 2).

Da jedes Mitochondrium 2-10 mtDNA-Kopien enthält und eine Zelle bis zu 10000 Mitochondrien hat, enthält eine Zelle Tausende von mtDNA-Kopien (Polyplasmie). Da die mtDNA nur 37 Gene enthält, von denen 24 für die Übersetzung der mtDNA benötigt werden, werden die meisten der etwa 1000 mitochondrialen Genprodukte von der nDNA kodiert und aus dem Zytosol importiert. Die mtDNA ist für ihre Replikations-, Transkriptions-, Translations- und Reparaturenzyme auf die nDNA angewiesen. Darüber hinaus sind die Mitochondrien für die meisten Proteine, die für die mitochondriale Funktion erforderlich sind, auf den Zellkern angewiesen. Während ihrer Entwicklung teilen und vermehren sich die Mitochondrien unter der Kontrolle des Kerns.
„Unsere Daten zeigen, dass das Spike-Protein eine langfristige transkriptionelle Unterdrückung von Genen des Mitochondrien-Stoffwechsels bewirken kann.“ Zumindest in übergewichtigen Mäusen (https://pubmed.ncbi.nlm.nih.gov/37348737/).
Gene des Mitochondrien-Stoffwechsels…
Wieviele Stoffwechselgene hat das Mitochondrium noch?
„37 Gene enthält, von denen 24 für die Übersetzung der mtDNA benötigt werden“
37-24=13
Kann es sein, dass das Spike-Protein eines dieser 13 Gene bzw. eines der 37 Gene stört? Damit ist das Mitochondrium dann wohl ziemlich kaputt (in übergewichtigen Mäusen).
Houston, we have a problem!!!!
Ich habe mir bereits vor einem Jahr folgende Frage gestellt, ob Mitochondrien von der Plasmidverunreinigung in den COVID-Spritzen transfiziert werden könnte. Zur Erinnerung, Plasmide sind zirkuläre DNA. Genauso zirkulär, wie das Genom der Mitochondrien. Ob da ein rundes Genom mehr auffallen würde?
https://drbine.substack.com/p/drbines-wilde-hypothese-konnte-das
Mitochondrien können transfiziert werden, zumindest durch Elektroporation (https://pubmed.ncbi.nlm.nih.gov/9030609/), möglicherweise auch durch Lipidnanopartikel.
In Arabidopsis thaliana (Ackerschmalwand, ein Unkraut) reichte für den Import eines Proteins in die Mitochondrien die Kombination aus einem auf Mitochondrien abzielenden Peptid und einem in die Zellen eindringenden Peptid. Die Sequenzen, die dafür gebraucht werden, werden schon sehr lange erforscht (https://www.sciencedirect.com/science/article/abs/pii/S1569255809600059, https://pubmed.ncbi.nlm.nih.gov/31696703/). Ob diese Sequenzen in den Covid-Injektionen enthalten sind, kann ich leider nicht sagen, das müsste jemand prüfen.
Die mtDNA ist GC-reich…
Was ist noch GC reich?
Genau!
Die codonoptimierten Sequenzen der Covid-Injektionen!
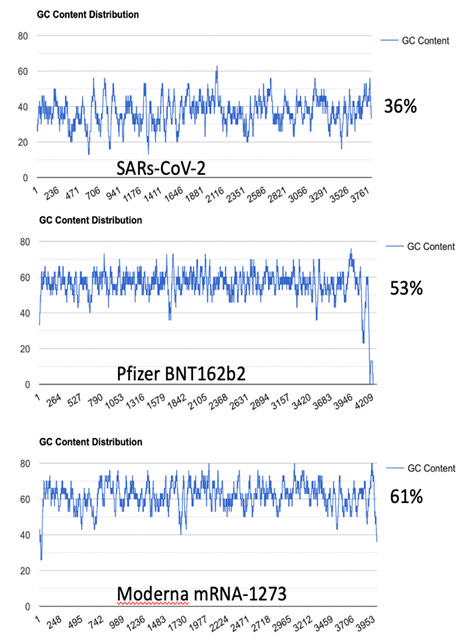
(https://doi.org/10.31219/osf.io/bcsa6)
Zufälle gibt’s aber auch.
Ganz abgesehen von den Problemen mit CpG Motiven (https://de.wikipedia.org/wiki/CpG-Dinukleotid)… (https://pubmed.ncbi.nlm.nih.gov/15532993/)…,
was für Proteine würden wohl vom Mitochondrium produziert, wenn es transfiziert würde, wenn man den alternativen genetischen Code des Mitochondriums zugrunde legt? Sicherlich nicht das gewünschte Spike-Protein sondern etwas ganz anderes, das wir aktuell weder kennen noch nachweisen können.
Kann das mal einer prüfen?
Gut für das Mitochondrium wird dieses Protein sicherlich nicht sein, würde ich mal vermuten.
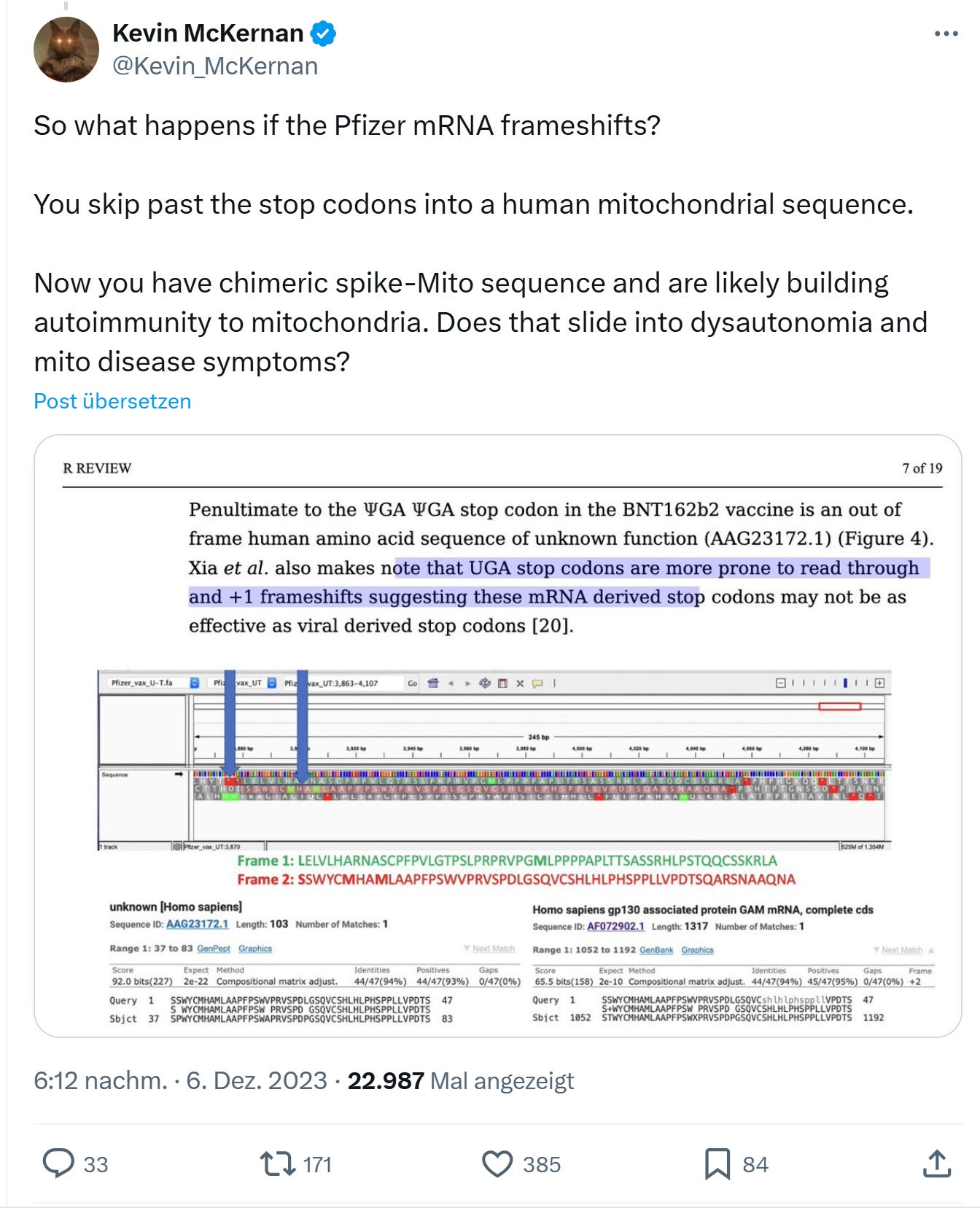
(https://x.com/Kevin_McKernan/status/1732447900360180035)
Was für ein “Zufall” dass UGA von Mitochondrien als Tryptophan gelesen wird…
Was für ein Protein würde in die Mitochondrien nach einem +1 frameshift wohl importiert?
Wir haben hier also ein Problem sowohl auf DNA/RNA Ebene als auch auf Proteinebene. Entweder könnte das Mitochondrium ein unbekanntes Protein produzieren, wenn es transfiziert würde bzw. es würde ein unbekanntes Protein unbekannter Funktion in die Mitochondrien importiert, gäbe es einen +1 frameshift.
Vielleicht passiert auch einfach beides. Doppelt genäht hält ja bekanntlich besser.
Grundlagen Mitochondriopathie
„Veränderungen in der mtDNA-Sequenz können vererbt oder somatisch (in situ entstanden) sein. Die Mutationsrate der mtDNA ist 10-20 Mal so hoch wie die der nDNA, was wahrscheinlich darauf zurückzuführen ist, dass das Korrekturlesen durch die mtDNA-Polymerasen nicht funktioniert. Normalerweise ist die gesamte mtDNA eines Individuums identisch (Homoplasmie).“
NORMALERWEISE sind die Probleme, die einer Mitochondriopathie zugrunde liegen, durch Mutationen in den mitochondrialen Genen, die vom Zellkern „betreut“ werden, verursacht.
Nun ist es aber so, dass es bei den Mitochondriopathien, welche den Covid-Injektionen zugrunde liegen, möglicherweise eine etwas andere Ursache gibt, die später ansetzt.
Meine Vermutung ist, dass das N1-Methylpseudouridin in die ribosomale RNA (rRNA) sowohl der zellulären Ribosomen als auch der mitochondrialen Ribosomen verbaut wird, dieser im 3D Raum verbiegt, so dass sich die Proteine der Ribosomen nicht mehr an das rRNA-Gerüst anlagern können.
Die komplette Herleitung gibt es in diesem Artikel:
Haben wir es teilweise vielleicht mit einer Ribosomopathie zu tun?

Mit diesem Artikel versuche ich meine Gedanken zu sortieren. Wenn jemand Ideen hat oder Informationen, die ich nicht kenne, einfach kommentieren. Ich habe das Huaier Paper[1] bisher nur am Rande wahrgenommen, da ich kein Arzt bin und Behandlung nicht meine Baustelle war und ist. Ich habe nur zur Kenntnis genommen, dass dieser Pilz namens Huaier das Probl…
Wenn also die 10.000 Proteine nicht mehr korrekt an das Mitochondrium geliefert werden, z. Bsp. verbogen sind, werden sie nicht mehr durch die Transporterproteine für den Import passen, bzw. wenn sie durchpassen, möglicherweise nicht mehr funktionieren, bzw. die Transporterproteine selbst verbogen/kaputt geliefert werden.
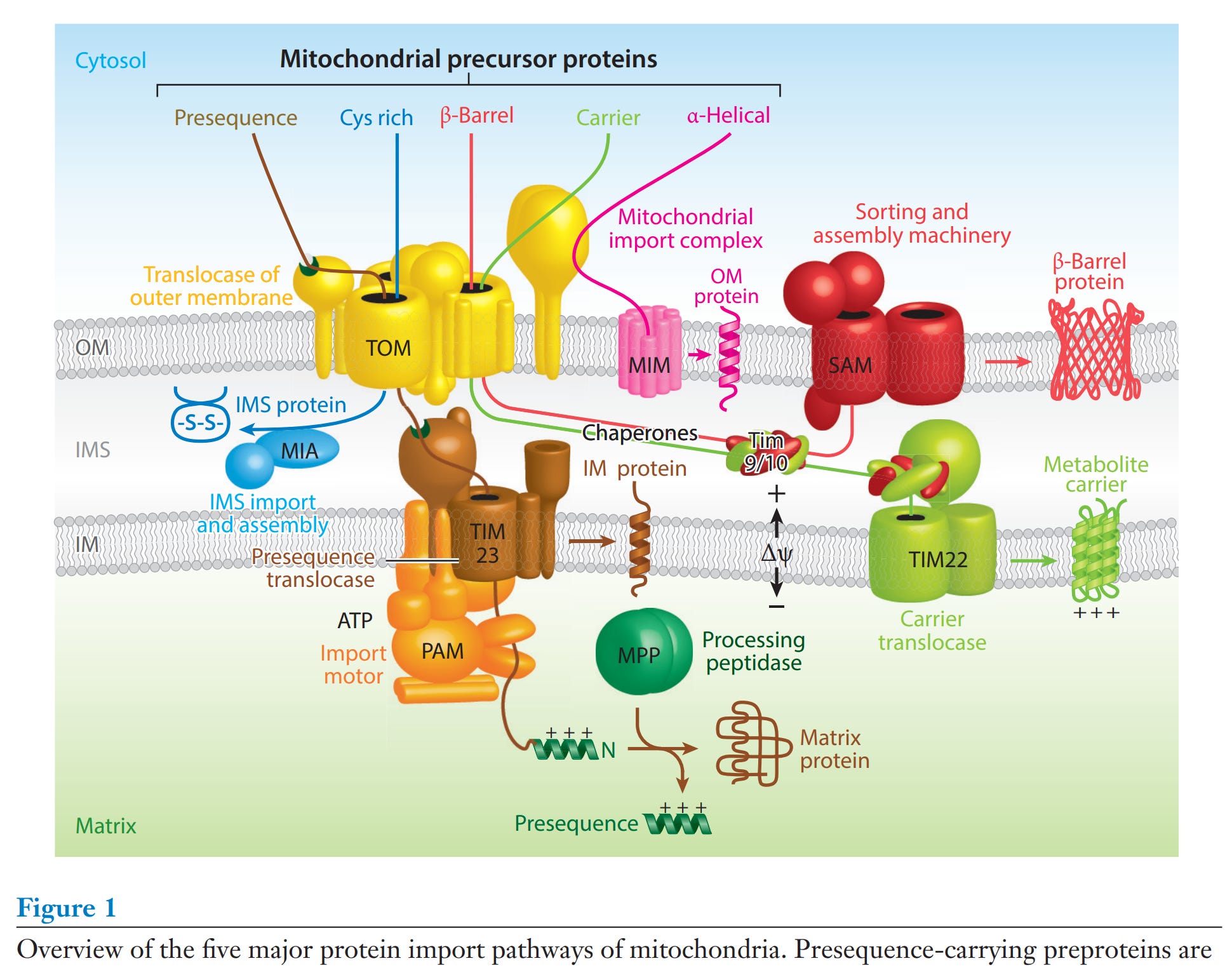
(https://pubmed.ncbi.nlm.nih.gov/28301740/).
Diese Proteinimportwege sehen komplex aus, sind komplex und die Proteine hierfür gehören zu den 10.000 Proteinen, die den Mitochondrien zugeliefert werden. Wenn da was schiefgeht… Das schaut mir nicht gut aus.
„Mitochondriopathien (MCP) sind entweder auf sporadische oder vererbte Mutationen in Genen zurückzuführen, die in der nukleären oder mitochondrialen DNA lokalisiert sind (primäre MCP), oder auf exogene Faktoren (sekundäre MCP).“
Symptome einer Mitochondriopathie
„Mitochondriopathien haben in der Regel einen chronischen, langsam fortschreitenden Verlauf und zeigen eine Multiorganbeteiligung mit unterschiedlichem Beginn zwischen der Geburt und dem späten Erwachsenenalter. Obwohl bei Mitochondriopathien mehrere Proteine mit Signal-, Aufbau-, Transport- und Enzymfunktionen beeinträchtigt sein können, ist in den meisten Fällen die Aktivität der Proteinkomplexe der Atmungskette (RC) primär oder sekundär betroffen, was zu einer beeinträchtigten Sauerstoffverwertung und reduzierten Energieproduktion führt.“
Ich erinnere in diesem Zusammenhang an dieses Paper:
"Die Ergebnisse, die für normale und Tumor-Gliazellen des menschlichen Gehirns (Astrozyten, Astrozytome, Glioblastome) erzielt wurden, die mit dem Covid-19 mRNA-Impfstoff von Pfizer/BioNT inkubiert wurden, zeigen Veränderungen in den Reduktions-Oxidationswegen, die mit Cytochrom c verbunden sind.“ (https://www.biorxiv.org/content/10.1101/2022.03.02.482639v1)
Cytochrom C ist Teil der Atmungskette, das habe ich in den Grundlagen ausführlichst erklärt. Cytochrom C ist auch an der Apoptose von beschädigten Zellen beteiligt. Cytochrom C scheint von diesem modRNA-Produkten oder vom Spike-Protein geschädigt zu sein.
„Mitochondriopathien stellen eine diagnostische Herausforderung dar, da sie sich in Präsentation und Verlauf stark unterscheiden. Häufig betroffene Systeme bei Mitochondriopathien sind
das periphere Nervensystem (Myopathie, Polyneuropathie, Laktazidose),
das Gehirn (Leukenzephalopathie, Verkalkungen, schlaganfallähnliche Episoden, Atrophie mit Demenz, Epilepsie, Zeichen der oberen Motoneuronen, Ataxie, extrapyramidale Manifestationen, Müdigkeit),
das Endokrinium (Kleinwuchs, Hyperhidrose, Diabetes, Hyperlipidämie, Hypogonadismus, Amenorrhoe, verzögerte Pubertät),
Herz (Defekte der Impulserzeugung oder -leitung, Kardiomyopathie, linksventrikuläre Herzinsuffizienz ohne Verdichtung),
Augen (Katarakt, Glaukom, pigmentäre Retinopathie, Optikusatrophie),
Ohren (Taubheit, Tinnitus, peripherer Schwindel),
Darm (Dysphagie, Erbrechen, Durchfall, Hepatopathie, Pseudoobstruktion, Pankreatitis, Pankreasinsuffizienz),
Niere (Nierenversagen, Zysten) und
Knochenmark (sideroblastische Anämie).“
Ich spare mit an dieser Stelle diese Symptome mit VAERS oder den PSURS zu vergleichen, die Überschneidungen sind mehr als offensichtlich. Daher auch der Disclaimer am Anfang.
„Abgesehen von den bekannten Syndromen sollte Mitochondriopathie bei jedem Patienten mit einer unerklärlichen progressiven Multisystemstörung in Betracht gezogen werden. Obwohl es eigentlich keine spezifische Therapie und Heilung für MCP gibt, erfordern viele sekundäre Probleme eine spezifische Behandlung. Das rasch wachsende Verständnis der pathophysiologischen Hintergründe der MCP könnte die Diagnose weiter erleichtern und Perspektiven für künftige, möglicherweise ursächliche Therapien eröffnen.“
„Mitochondriopathien (MCP) sind entweder auf sporadische oder spontane Mutationen der mitochondrialen DNA (mtDNA) oder der Kern-DNA (nDNA) in Genen zurückzuführen, die für Enzyme, Struktur-, Signal-, Träger-/Shuttle-, Kanal-, Rezeptor-, Hitzeschock- oder Assemblierungsproteine, tRNAs oder rRNAs des Mitochondriums kodieren, oder auf exogene Noxen wie Medikamente, Toxine oder Infektionen. Die am häufigsten von Mutationen betroffenen Proteine sind die der Atmungskette (RC) und der oxidativen Phosphorylierung (OXPHOS). Deshalb werden Mitochondriopathien oft synonym als Atmungskettenstörungen (RCDs) bezeichnet. Mitochondriopathien können jedoch auch auf Defekte in anderen Bahnen und Komponenten des Mitochondriums als der Atmungskette oder der oxidativen Phosphorylierung zurückzuführen sein. Mitochondriopathien weisen ein breites Krankheitsspektrum auf und ihre klinischen Merkmale überschneiden sich. Bei der mtDNA kann eine einzige Mutation oder verschiedene Mutationen in demselben Gen unterschiedliche klinische Manifestationen aufweisen (Phänokopie), während derselbe klinische Phänotyp durch verschiedene Mutationen verursacht werden kann (genetische Heterogenität).“
Liste bekannter Mitochondriopatien
Die Liste der bekannten Mitochondriopathien war schon 2004 richtig lang und gut sortiert.
Man kann sie genetisch sortieren:

Man kann sie biochemisch sortieren:

Man kann sie klinisch sortieren und ihnen hübsche Syndromnamen geben, obwohl man die Ursache ja kennt, und es somit eigentlich kein waschechtes Syndrom ist.

Und erneut, viele alte Bekannte.
Man kann sie nach häufigen Manifestationen sortieren.
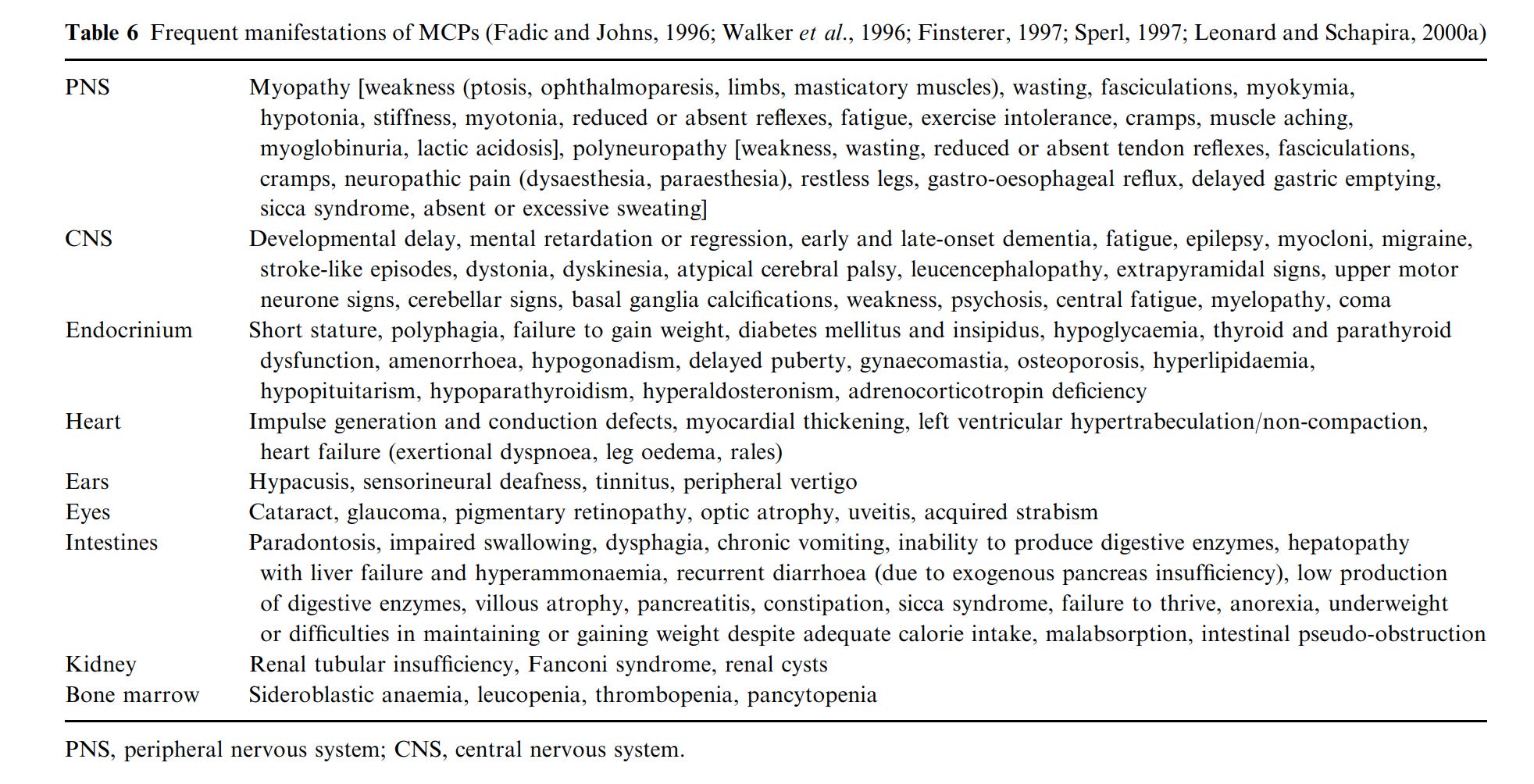
Und erneut, viele alte Bekannte. Besonders bekannt die PNS Symptome mit fatigue, exercise intolerance, neuropathy, restless legs… Aber auch die anderen Symptome, alles gute, alte PostVac Symptome. Aber, ich erinnere erneut an den Disclaimer am Anfang. Das kann man auch alles mit anderen Mechanismen erklären. Die Überschneidungen sind aber… auffällig.
Teilweise kennt man exakt die betroffenen Gene, was man sich im Kontext der modRNA sparen kann, weil, wie erwähnt, kann auf Proteinebene passieren und muss nicht ein Gen im Zellkern sein, das natürlich auch, durch Insertion der modRNA in den Kern betroffen sein kann, aber dann nicht in dem Ausmaß, wie man es beobachtet.
Das Paper hat Tipps in Sachen Diagnostik, aber da hat die Diagnostik mittlerweile schon ein wenig Fortschritte gemacht, daher spare ich mir diesen Teil, da gibt es mittlerweile andere Möglichkeiten.
Bei der Liste der möglichen Therapien könnte vielleicht das eine oder andere Nützliche dabei sein.

Und auch hier, viele alte Bekannte aus dem Bereich der Nahrungsergänzungsmittel der PostVac Behandlung. Sicherlich nur Zufall, dass die auch bei PostVac funktionieren.
Fazit:
Es gibt mehrere Mechanismen der modRNA-Produkte, die zu einer Mitochondriopathie führen können:
N1-Methylpseudouridine zerstört durch Fehleinbau die ribosomale RNA sowohl der zellulären als auch mitochondrialen ribosomalen RNA und somit auch die Ribosomen und somit die Proteinproduktion.
Das Spike-Protein zerstört die Atmungskette.
Deregulierung der RAS
Autoimmunangriff auf die Mitochondrien.
Die Symptome sind alte Bekannte, die auch durch andere Mechanismen verursacht werden, wie cGAS/STING, RAS, Endotoxine, Autoimmunreaktionen…
Des Weiteren können gestresste Mitochondrien zu Krebs, Entzündungen oder Thrombosen führen.
Und bei all dem geht es “nur” im die Mitochondrien, die in den Zellen wohnen. Da sind wir noch nicht bei den Freischwimmern und deren möglichen Funktionen:
Freischwimmende Mitochondrien und freischwimmende DNA

Es sieht so aus, als wenn in unserem Blut freie Mitochondrien herumschwimmen, die nicht in Zellen leben (Scientists Find Cell-Free Mitochondria in Human Blood | Sci.News). Diese Mitochondrien haben eine aktive Atmungskette und produzieren Energie. „Sowohl das Plasma gesunder Personen als auch Zellkulturmedien enthalten strukturell intakte Mitochondrien. …
„In vitro führen nur 1-2 % der durch Lipidnanopartikel (LNP) vermittelten Nukleinsäureübertragung zu einer erfolgreichen Zelltransfektion. Das pathogene Potenzial der restlichen 98 % ist jedoch noch nicht ausreichend erforscht.“ (https://pubmed.ncbi.nlm.nih.gov/38263456/)
Was wäre wenn die freischwimmenden Mitochondrien transfiziert würden, fremdes Protein herstellten und dann durch einen Autoimmunangriff getötet würden? Da wir nicht wissen, was für ein Protein von den Mitochondrien hergestellt würde, können wir darauf aktuell nicht testen.
Wie groß ist der Anteil der freischwimmenden Mitochondrien an der Energieproduktion des Körpers?
Würde das Immunsystem dadurch auf Mitochondrien geprimed?
DoorlessCarp hat sich des Mitochondrienproblems von einer etwas anderen Seite angenommen, sieht genauso schlimm aus, nur mit anderen Schwerpunkten.

Updates:
20.09.2024: Das Spike Protein selbst schädigt die Mitochondrien
Wenn ich es schaffe diesen Artikel komplett aufzufassen und wiederzugeben(ich bin noch lange nicht soweit), kann ich glaube ich auch das Studium in Biologie anfangen mit meinen 42 Jahren 😁🫶 ich danke ihnen vom Herzen, für diese wunderbare Arbeit. So eine Arbeit verdient so viel mehr Liebe.
N1-Methylpseudouridine destroys the ribosomal RNA of both the cellular and mitochondrial ribosomal RNA through incorrect incorporation and thus also the ribosomes and thus protein production
Yes, I've wondered about this from the very beginning.
THanks