


Deregulation von praktisch allem über RAS und AT1R
Systembiologie vom Feinsten

Disclaimer und Spoiler:
Es mag nach diesem Artikel vielleicht so klingen, dass die RAS-Kaskade die Ursache für alle Nebenwirkungen sein könnte. ABER VORSICHT! Der Körper hat N Möglichkeiten sich zu äußern. Diesen Symptomen können aber X verschiedene Ursachen zugrunde liegen.
Viele Autoren erliegen leider der Versuchung zu glauben, sie hätten die Ursache gefunden, die man behandeln muss und das Problem ist gelöst. Es gibt aber zu viele Mechanismen da draußen, die alle jeweils alleine die Symptome erklären könnten. Beispiele wären cGAS/STING, Endotoxine, Mitochondriopathie, MCAS, RAS… ABER, wenn diese Mechanismen parallel/gleichzeitig aktiv sind, wird es immer noch nur die N möglichen Symptome geben, denen X verschiedene Schäden zugrunde liegen. Mitochondriopathie kann tatsächlich über durch RAS losgetretene ROS mit entstehen, was es nicht einfacher macht.
Daher, lieber Leser, RAS ist erneut nur ein weiterer Mechanismus von vielen, die ausgelöst werden und nicht die alleinige Ursache, die es zu behandeln gilt, nur eine der Ursachen, eine der vielen Ursachen, die alle bekannten Symptomatiken bereits alleine erzeugen kann.
Und ja, dieser Disclaimer ist copy und paste von Mitochondriopathie und MCAS mit minimalen Anpassungen und ich befürchte, ich werde ihn noch öfter brauchen.
Die RAS-Kaskade läuft auch unter RAAS-Kaskade. Die Grundlagen hatte ich dazu bereits in diesem Substack angeschnitten: https://drbine.substack.com/p/schadmechanismen-durch-bindungblockade
Jean-Marc Sabatier ist jedoch DEUTLICH tiefer in das Thema eingestiegen und kann in seinem Paper
El-Arif G, Khazaal S, Farhat A, Harb J, Annweiler C, Wu Y, Cao Z, Kovacic H, Abi Khattar Z, Fajloun Z, Sabatier JM. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules. 2022 Mar 22;27(7):2048. doi: 10.3390/molecules27072048. PMID: 35408447; PMCID: PMC9000463. https://pubmed.ncbi.nlm.nih.gov/35408447/
unter anderem herleiten, wie das Spike-Protein über Ras das Immunsystem dereguliert (und noch diversen anderen Schabernack treibt).
Dieses Paper dient als Grundgerüst für diesen Substack. Ich werde daher die Stellen, die auf diesem Sabatier Paper beruhen nicht explizit referenzieren, auch wenn ich nach ihnen paraphrasiere.
DAS WIRD EIN HARTER RITT durch Signalkaskaden unter dem selbst ich gelitten habe. Ich werde versuchen die Grundlagen, die man braucht, um das Paper aus der Sabatier Gruppe zu verstehen, in diesem Substack jeweils nachzufüttern. Ich gehe davon aus, wenn ich die Infos nachschlagen muss, muss ich sie auch nachfüttern, weil sie dir, geneigter Leser, auch fehlen werden.
Ich hasse Pathways.
Ich hasse Pathways mit Abzweigungen.
Ich hasse es, wenn Pathways diverse andere Pathways lostreten.
All das ist hier leider der Fall.
Der französische Autor Jean-Marc Sabatier ist in Frankreich ziemlich bekannt und schreibt z. Bsp. für France Soir (https://www.francesoir.fr/recherche?search_api_fulltext=Jean-Marc+Sabatier) oder infodujour (https://infodujour.fr/?s=Jean-Marc+Sabatier). In Deutschland oder im englischsprachigen Umfeld habe ich nichts von ihm gehört, er ist aber in Frankreich durchaus eine große Nummer (https://www.scholarena.com/eb/308/Jean-Marc-Sabatier-Journal-of-Neuroscience-and-Neuropsychology) und hat zum Thema Spike-Protein und RAS-Kaskade zwei Bücher veröffentlicht (https://www.amazon.de/dp/2813231185/ und https://www.amazon.de/COVID-19-articles-censur%C3%A9s-Jean-Marc-Sabatier/dp/B0BSJFZC4T/).
Die Wirkung des SARS-CoV-2-Virus und des SARS-CoV-2-Spike-Proteins auf das RAS-System sind gleichzusetzen, da das SARS-CoV-2-Virus über das Spike-Protein an das Angiotensin-konvertierende Enzym 2 (ACE2) bindet. Das RAS unterscheidet nicht, ob am Spike-Protein noch ein Virus dran hängt oder nicht. Schlüssel-Schloss-Prinzip, Mittelstufenschulstoff. Das Spike-Protein bindet an ACE2, egal ob es Teil des Virus ist oder nicht.
Dass das Spike-Protein an ACE2 bindet, bestreitet keiner. BioNTech/Pfizer haben das sogar selbst in ihren Daten gezeigt (https://phmpt.org/wp-content/uploads/2023/02/125742_S1_M4_4.2.1-vr-vtr-10741.pdf Seite 10) und das Prolin-Schloss hat nicht funktioniert, das genau das verhindern sollte (https://pubmed.ncbi.nlm.nih.gov/35994646/).
Kurzversion (Take Home Message für Anwälte)
Die Bindung des Spike Proteins an ACE2 führt zu einer Gleichgewichtsverschiebung im für die Regulation von Körperfunktionen extrem wichtigen und zentralen Renin-Angiotensin System (RAS), was zu eine Überschuss von Ang II führt, welches den gefäßverengenden Rezeptor vom Typ 1 (AT1R) aktiviert.
Der gefäßverengenden Rezeptor vom Typ 1 (AT1R) aktiviert seinerseits DIVERSE Signalkaskaden, die ihrerseits komplexe MEHRSTUFIGE Signalkaskaden aktivieren.
p38/MAPK (Die p38-Familie ist eine evolutionär konservierte Gruppe von mitogen-aktivierten Proteinkinasen (MAPKs), die an der Koordinierung zellulärer Reaktionen auf fast alle Stressreize beteiligt ist und dazu beiträgt. (https://pubmed.ncbi.nlm.nih.gov/32612808/) Der MAP-Kinase-Weg besteht aus einer Reihe mehrstufiger Signaltransduktionswege.
NADPH-Oxidase (Nox) führt zur Produktion von reaktiven Sauerstoffspezies (ROS), die Schaden anrichten
NF-κB, welches von großer Bedeutung für die Regulation von Immunantwort, Zellproliferation, Zelltod, Entzündungen und Entwicklung des Immunsystems ist. (https://de.wikipedia.org/wiki/NF-%CE%BAB).
JAK/STAT3-Signalweg, der spielt bei verschiedenen Krebsarten eine wesentliche Rolle
Kann zu einem M1/M2 Makrophagen Shift führen
Proto-Oncogene Mas Receptor (MASR) Aktivierung, d.h. RAS ist an der (Bust-)Krebsentstehung beteiligt (https://www.mdpi.com/2073-4409/9/6/1336)
…

Wir haben es mit einem extrem komplexen systembiologischen Problem zu tun, welches (mit an Sicherheit grenzender Wahrscheinlichkeit) noch Unbekannte Unbekannte enthalten könnte.
Und man wusste von Anfang an, dass das Spike-Protein an ACE2 bindet.
Man kannte das Renin-Angiotensin System (RAS).
Man ist dabei, die Pathophysiologie des RAS zu erforschen und hat gerade neue Komponenten entdeckt (Alamandine (hat cardioprotektive Eigenschaften) und MrgD), die Sabatier noch gar nicht auf dem Schirm hat und die daher hier noch gar nicht behandelt werden (https://pubmed.ncbi.nlm.nih.gov/37350982/)).
Man hat die Menschen dennoch dazu gebracht ein Protein in unbekannter Menge für unbekannte Zeit zu produzieren, von dem man wusste, dass es über Bindung an ACE2 RAS deregulieren wurde.
Man hat die damit losgetretenen, unübersichtlichen, unvorhersagbaren Kaskaden billigend in Kauf genommen.
Viele Organe besitzen ACE2 an ihrer Oberfläche (https://www.proteinatlas.org/ENSG00000130234-ACE2/tissue).
Die Schäden, die dadurch entstehen umfassen unter anderem:

Der Angiotensin II Type I Rezeptor (AT1R) befindet sich zudem auch an der Oberfläche vieler Immunzellen, einschließlich T-Zellen. Diese Immunzellen besitzen somit eine zelleigene, endogene RAS-Signalkette und produzieren so selbst Angiotensin II. Angiotension II fördert in Immunzellen die Proliferation (Zellteilung), Migration, Differenzierung, Adhäsion und Effektorfunktion der Immunzellen und fungiert als costimulatorisches Molekül, das für die Aktivierung von T-Zellen wichtig ist.
Die Effektorwirkungen der Immunzellen werden durch die AT1R-Bindung ausgelöst. Dies führt zu einer erhöhten Produktion von proinflammatorischen Zytokinen durch CD4+ T-Zellen (T-Helferzellen) und Perforin durch CD8+ T-Zellen (T-Killerzellen) und somit einer Deregulierung in den internen Signalkaskaden der Immunzellen und somit des Immunsystems.
Ein Beleg für Sabatiers RAS-Hypothese ist, dass Sartane also angiotensin II type 1 receptor blockers (ARBs) (Candesartan or Telmisartan) für 6 Wochen “seltsamer Weise” bei Post-Covid helfen. (https://www.mdpi.com/1422-0067/25/8/4522)
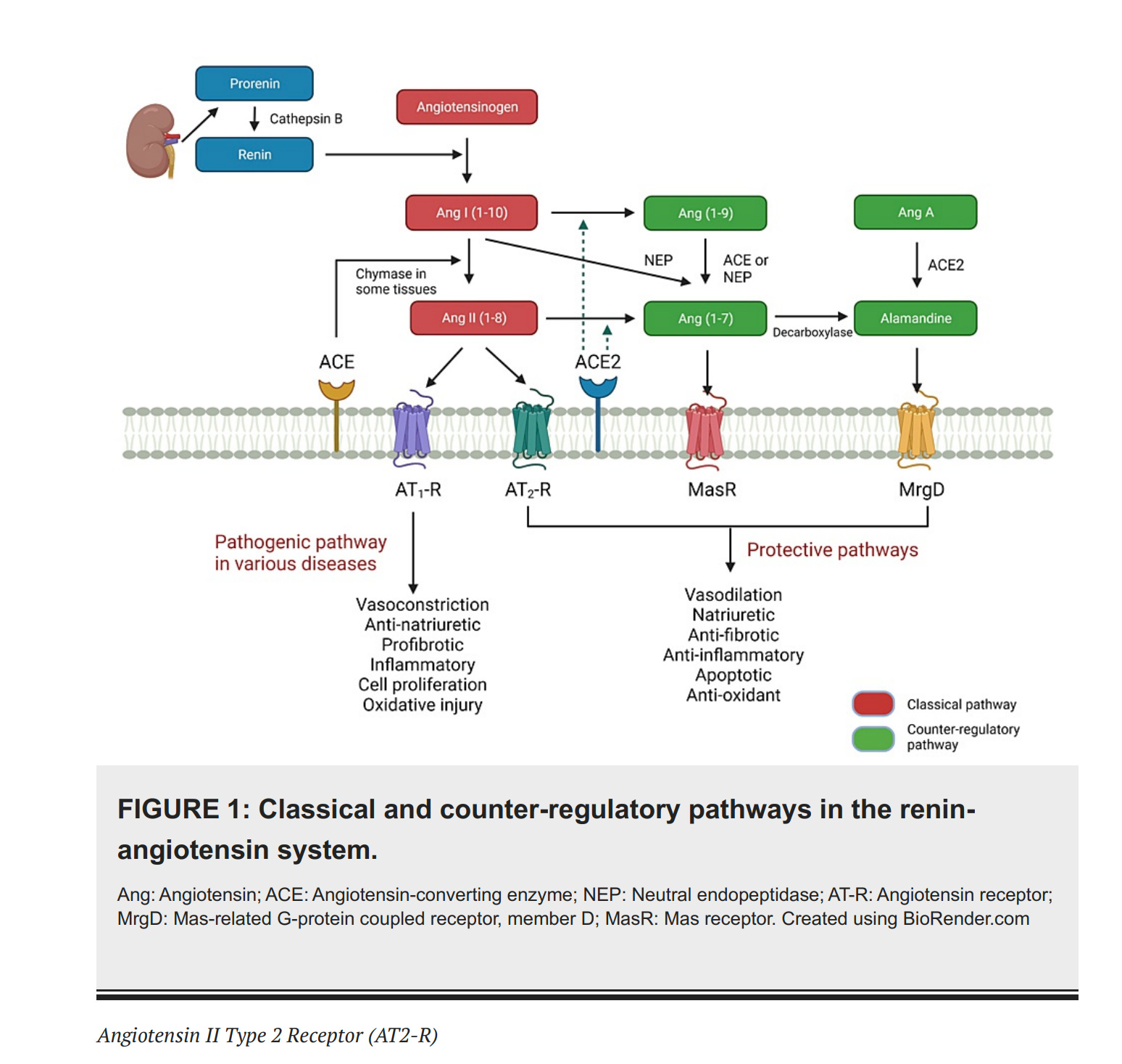
Die Renin-Angiotensin System (RAS): Grundlagen
Der Zweck des Renin-Angiotensin System (RAS) ist, das von der Leber ausgeschüttete Angiotensionogen (AGT) mit dem von der Niere ausgeschütteten Enzym Renin zu spalten, um so Angiotension I (Ang I) zu produzieren.
Die Niere schüttet das Enzym Renin aus. Renin, weil „von der Niere“ als renal bezeichnet wird. Renin ist das Substantiv des Adjektivs renal.
Die Leber schüttet Angiotensinogen (AGT) aus. Angiotensinogen ist ein Protein und Prohormon.
Das Enzym Renin aus der Niere spaltet das Angiotensinogen (AGT) aus der Leber in das 10 Aminosäuren kleine Peptid Angiotensin I (Ang I).
Angiotensin I (Ang I) wird durch das Angiotensin-konvertierende Enzym (ACE) gespalten. Dieses Spaltprodukt nennt man Angiotensin II (Ang II).
Angiotensin II (Ang II) bindet sowohl an den gefäßverengenden Rezeptor vom Typ 1 (AT1R) als auch an gefäßerweiternde Rezeptoren vom Typ 2 (AT2R).
Angiotensin II (Ang II) wird auch durch das Angiotensin-konvertierende Enzym 2 (ACE2) gespalten, um so Angiotensin (1-7) [Ang-(1-7)] zu erzeugen.
Angiotensin (1-7) [Ang-(1-7)] interagiert mit dem proto-onkogenen G-Protein-gekoppelten Rezeptor Mas (MasR) und dem gefäßerweiternde Rezeptoren vom Typ 2 (AT2R) interagiert.
Das Peptid Angiotensin II (Ang II), übt mehrere schädliche Wirkungen aus, wenn es an den gefäßverengenden Rezeptor vom Typ 1 (AT1R) bindet
Vasokonstriktion
Profibrose
Proapoptose
oxidativen Stress
Proinflammation.
Das Angiotensin-konvertierende Enzym 2 (ACE2) gleicht die Wirkung des Bindung von Angiotensin II (Ang II) an den gefäßverengenden Rezeptor vom Typ 1 (AT1R) aus, wirkt also als eine Art Gegengewicht. Das macht ACE2 indem es Ang I und Ang II in Angiotensin (1-9) [Ang-(1-9)] bzw. Ang-(1-7) spaltet.
Wenn das Spike-Protein mit dem Angiotensin-konvertierende Enzym 2 (ACE2) interagiert, also an ACE2 bindet, welches der Rezeptor für das Spike-Protein ist, obwohl es auch ein Enzym ist, führt das zu einem Ungleichgewicht im RAS und zu einer Überaktivierung des gefäßverengenden Rezeptor vom Typ 1 (AT1R).

(https://pubmed.ncbi.nlm.nih.gov/37711109/)
Das passiert in allen Zellen, die ACE2 an der Zelloberfläche haben und das sind viele Zellen siehe (https://drbine.substack.com/p/schadmechanismen-durch-bindungblockade).
DAS ist Schadensstufe 1, also “nur” Schäden durch die RAS Deregulierung.
Die RAS-Kaskade und das Immunsystem
Schadensstufe 2 wird durch das deregulierte RAS über AT1R losgetreten.
Es hat sich gezeigt, dass ACE–Angiotensin II (Ang II) nicht nur bei Blutdruck und Wasserhaushalt eine Rolle spielt, sondern auch bei der Regulierung des Immunsystems.
Der Angiotensin II Type I Rezeptor (AT1R) befindet sich an der Oberfläche vieler Immunzellen, einschließlich T-Zellen. Diese Immunzellen besitzen somit eine zelleigene, endogene RAS-Signalkette und produzieren so selbst Angiotensin II. Angiotension II fördert in Immunzellen die Proliferation (Zellteilung), Migration, Differenzierung, Adhäsion und Effektorfunktion der Immunzellen und fungiert als costimulatorisches Molekül, das für die Aktivierung von T-Zellen wichtig ist.
Die Effektorwirkungen der Immunzellen werden durch die AT1R-Bindung ausgelöst. Dies führt zu einer erhöhten Produktion von proinflammatorischen Zytokinen durch CD4+ T-Zellen (T-Helferzellen) und Perforin durch CD8+ T-Zellen (T-Killerzellen) sowie zu einer erhöhten Adhäsions- und Migrationsfähigkeit durch Hochregulierung von Adhäsionsmolekülen und Chemokinrezeptoren.

Oder ein wenig biochemischer als bei Sabatier:

(https://pubmed.ncbi.nlm.nih.gov/35027697/). In diesem Schaubild hängt das Spike-Protein noch am Virus, geht aber ohne Virus. Mit Virus am Spike bekommt man die Daten einfacher publiziert.
Wenn das Spike-Protein mit dem Angiotensin-konvertierende Enzym 2 (ACE2) interagiert, führt das zu einem Ungleichgewicht im RAS und zu einer Überaktivierung des gefäßverengenden Rezeptor vom Typ 1 (AT1R).
Die Bindung des Pike-Proteins an ACE2 über Schlüssel-Schloss-Prinzip reguliert das Enzym ACE2 herunter. Das herunterregulieren von ACE2 führt zu einem Anstieg des Ang II-Spiegels, weil ACE2 kein Ang (1-7) und kein Ang (1-9) mehr herstellt, daher kann mehr Ang II entstehen.
Ang II bindet an den gefäßverengenden Rezeptor vom Typ 1 (AT1R).
AT1R wiederum aktiviert eine Reihe von Signalwegen wie PKC und des ERK-Signalwegs, die an der Aufrechterhaltung der Kontraktion und des Zellwachstums beteiligt ist. (https://pubmed.ncbi.nlm.nih.gov/16870827/).
Angiotensin II (Ang II) erhöht die Produktion von reaktiven Sauerstoffspezies (reactive oxygene species ROS)-Produktion durch AT1R-abhängige Induktion von NADPH-Oxidase (Nox), was zu mitochondrialer Dysfunktion und DNA-Schäden führt und die Induktion von Entzündungszytokinen durch NF-κB unterstützt.

(https://pubmed.ncbi.nlm.nih.gov/16870827/)
Hier etwas genauer, wie das mit Nox läuft. Nox ist wieder einmal eine ganze Familie von Proteinen:

Die wichtigste Quelle für ROS im kardiovaskulären System ist die NOX-Familie (NADPH-Oxidase). NOX1-4 sind p22phox-abhängige Oxidasen, während NOX5, ein Ca2+-empfindliches NOX, für seine Aktivierung kein p22phox benötigt. Im kardiovaskulären System werden NOXe durch blutdrucksenkende und entzündungsfördernde Faktoren reguliert, darunter Ang II (Angiotensin II), ET-1 (Endothelin-1), Aldosteron, Salz, Wachstumsfaktoren (VEGF [vascular endothelium growth factor] und epidermal growth factor [EGF]) und TNF (tumor necrosis factor). ROS werden auch durch die Entkopplung von eNOS (endotheliale Stickoxid-Synthase) sowie durch mitochondriale und endoplasmatische Retikulum-Mechanismen (ER) erzeugt, die durch NOX/ROS beeinflusst werden. AT1R steht für den Ang-II-Typ-1-Rezeptor, BH4 für Tetrahydrobiopterin, ETAR für den ET-1-Typ-A-Rezeptor, GFR für den Wachstumsfaktor-Rezeptor und MR für den Mineralocorticoid-Rezeptor.(https://pubmed.ncbi.nlm.nih.gov/33793335/)

(https://pubmed.ncbi.nlm.nih.gov/19390623/)
„NF-κB (nuclear factor 'kappa-light-chain-enhancer' of activated B-cells) ist ein spezifischer Transkriptionsfaktor, der in praktisch allen tierischen Zelltypen und Geweben vorkommt. Über die Bindung an bestimmte regulatorische Abschnitte der DNA kann er die Transkription abhängiger Gene beeinflussen.
NF-κB ist von großer Bedeutung für die Regulation der Immunantwort, der Zellproliferation und des Zelltodes. Die Aktivierung von NF-κB gilt als kritisch für die Entstehung von Entzündungen. Schließlich erfüllt NF-κB wichtige Funktionen im Bereich der Entwicklung des Immunsystems und der lymphatischen Organe. Die Rolle von NF-κB in anderen Zusammenhängen (z. B. im Nervensystem) ist Gegenstand gegenwärtiger Forschung.
Aufgrund seiner vielfältigen Funktionen wird NF-κB auch mit zahlreichen Erkrankungen in Zusammenhang gebracht. Dabei ist vielfach unklar, inwieweit die Aktivierung von NF-κB tatsächlich kausal in den Krankheitsprozess eingreift. Bei einigen Arten von Krebserkrankungen wird eine solche Rolle zunehmend als wahrscheinlich angesehen, so dass Bestandteile des NF-κB-Signalweges inzwischen wichtige Zielstrukturen für die Entwicklung neuer Medikamente geworden sind.“ (https://de.wikipedia.org/wiki/NF-%CE%BAB)
ADAM17 wird auch durch die Ang II-AT1R-Achse über die intrazelluläre p38/MAPK aktiviert und verdaut die Membranformen von EGF und TNF-α, wobei ihre löslichen Formen entstehen, die alle auch den NF-κB-Weg über ihre spezifischen Rezeptoren stimulieren.
„Der MAP-Kinase-Weg (MAP, englisch mitogen-activated protein) bezeichnet in der Biologie eine Reihe mehrstufiger Signaltransduktionswege, die unter anderem an der Regulation der Embryogenese, der Zelldifferenzierung, des Zellwachstums und des Programmierten Zelltodes beteiligt sind.“
Hier sind nur Verbindungen eingezeichnet. Hier und in allen anderen Schaubildern fehlen eigentlich feed-forward und feed-back loops, d.h. es ist eigentlich noch DEUTLICH komplexer. Ganz abgesehen davon, dass möglicherweise noch Proteine fehlen, die man noch gar nicht kennt.

(https://de.wikipedia.org/wiki/MAP-Kinase-Weg)

(https://elisa-kits.de/de/p38-mapk-signaling-pathway_de)
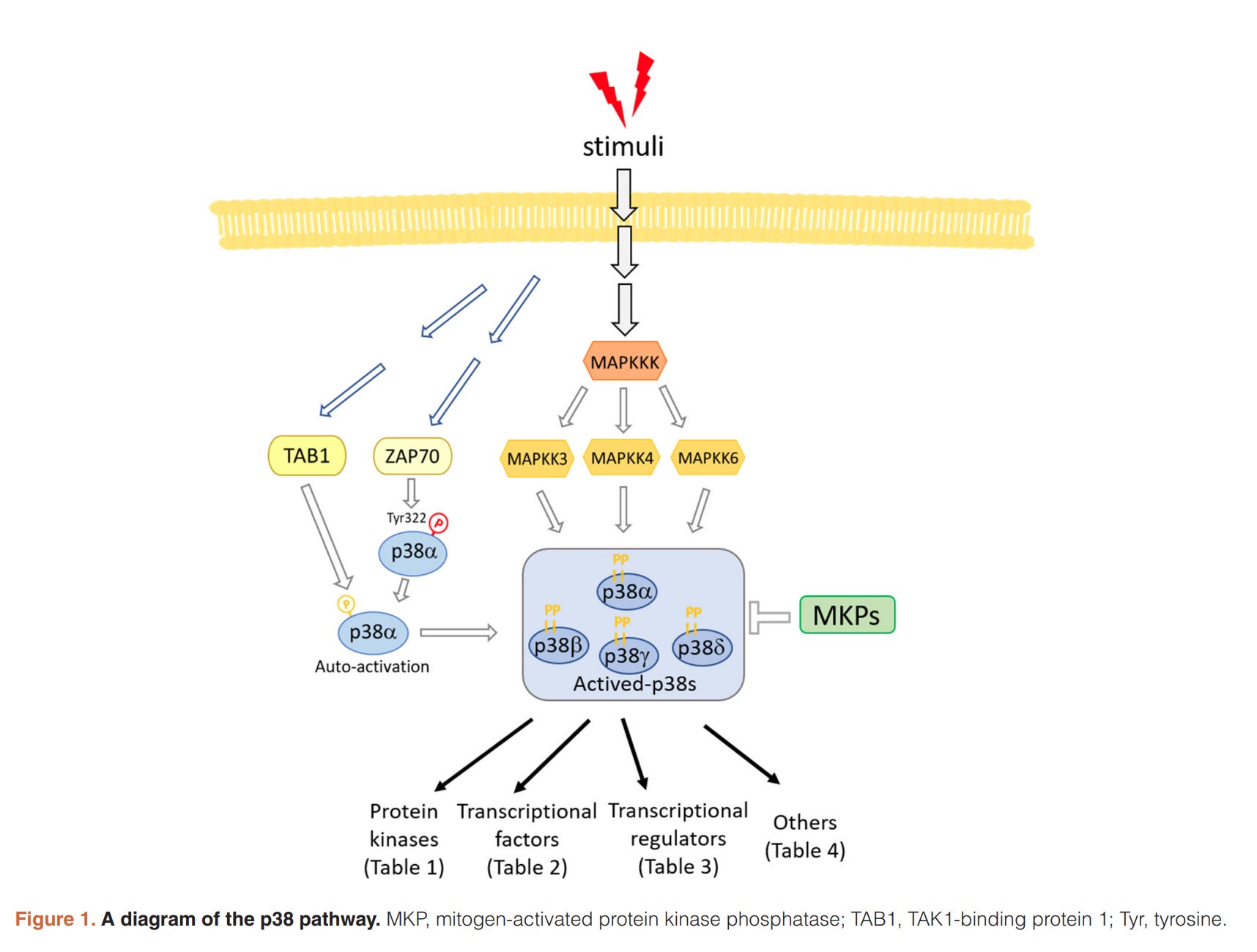
(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7324945/)

(https://pubmed.ncbi.nlm.nih.gov/32471115/)
EGF (Synonyme: epidermaler Wachstumsfaktorrezeptor, ErbB1, HER1) Die wichtigsten beiden Signalwege sind die bekannten Ras-Raf-MEK-ERK- sowie der PI3K-AKT-mTOR-Signalweg. Des Weiteren findet eine Aktivierung von STAT3/5 und Proteinkinase C statt. Das so weitergereichte Signal stimuliert letztendlich das Zellwachstum, verhindert den apoptotischen Zelltod und trägt somit zur Transformation der Zelle bei. […] Man findet eine deutliche Überexpression des Rezeptors in vielen nicht-kleinzelligen Bronchialkarzinomen, Glioblastomen, Nierenzellkarzinomen, Ovarialtumoren sowie Colonkarzinomen und anderen Tumorentitäten. Mutationen sind in vielen der insgesamt 28 Exons, sowie in den Intronregionen beschrieben.“ (https://flexikon.doccheck.com/de/EGF-Rezeptor)
TNF-α (Synonyme: Tumornekrosefaktor-α, TNF-α, TNF alpha, TNFA, Kachexin)
Der Tumornekrosefaktor wird hauptsächlich von Makrophagen, jedoch zu einem geringen Anteil auch von Mastzellen, Lymphozyten, Endothelzellen, Herzmuskelzellen, Fibroblasten und Nervenzellen ausgeschüttet. Auch Fettzellen (Lipozyten) können den Tumornekrosefaktor sezernieren, weshalb er auch den so genannten Adipokinen zugerechnet wird. Der Tumornekrosefaktor hat verschiedene Wirkungen:
auf den Hypothalamus:
Ausschüttung von Corticotropin-releasing Hormon (CRH)
Appetitminderung
Auslösen von Fieber
auf die Leber:
Akute-Phase-Proteine werden vermehrt ausgeschüttet.
Insulinresistenz wird erhöht, indem Insulinrezeptoren phosphoryliert werden.
Chemotaxis von neutrophilen Granulozyten
auf Makrophagen: Stimuliert zur Phagozytose
auf anderes Gewebe: steigert die Insulinresistenz
Eine zu hohe Konzentration von TNF kann über einen längeren Zeitraum zur Kachexie (Abmagerung) führen. Dies kann bei einigen Tumorpatienten beobachtet werden. Eine Überproduktion von TNF wird auch bei einigen Tumorpatienten gefunden, weshalb es auch als sogenannter Tumormarker in der Diagnostik eingesetzt wird.“ (https://flexikon.doccheck.com/de/TNF-alpha)
Außerdem spaltet ADAM17 das membrangebundene IL-6Rα in die lösliche Form (sIL-6Rα), die einen Komplex mit IL-6 bildet und an gp130 bindet, was zur Aktivierung von JAK/STAT3 und zur Induktion von überschüssigem IL-6 führt.

(https://pubmed.ncbi.nlm.nih.gov/33014208/)
Der JAK/STAT3-Signalweg spielt bei verschiedenen Krebsarten eine wesentliche Rolle. Die Aktivierung dieses Signalwegs führt zu einer erhöhten tumorigenen und metastatischen Fähigkeit, zur Umwandlung von Krebsstammzellen (CSC) und zur Chemoresistenz bei Krebs, indem der epithelial-mesenchymale Übergang (EMT) gefördert wird.

(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7017057/)

(https://onlinelibrary.wiley.com/doi/10.1155/2017/5126048)
Als wäre dieses Systembiologische Pathwaychaos nicht schon schlimm unübersichtlich genug, kann spielt AT1R spielt auch noch eine wichtige Rolle bei der Regulierung des Makrophagen-Phänotyps, kann also einen M1, M2 Shift hervorrufen via Hypoxie-induzierbaren Faktor (HIF)1α, Toll-like-Rezeptor (TLR)4 und das Verhältnis von pIκB/IκB. (https://pubmed.ncbi.nlm.nih.gov/35721173/)
“Immunzellen, die als M1-Makrophagen bekannt sind, spielen eine Rolle bei der Auslösung bestimmter Immun- und Entzündungsreaktionen. M2-Makrophagen hingegen sind entzündungshemmende Zellen, die Effektor-T-Zellen durch die Freisetzung von Interleukin 10 (IL-10) hemmen.
Der M2-Phänotyp ist der vorherrschende Phänotyp bei tumorassoziierten Makrophagen und spielt eine wesentliche Rolle bei der Stimulierung von Tumorwachstum, Invasion und Metastasierung. Das Endothel kann anfälliger für die Invasion von Tumorzellen werden, wenn M2-Makrophagen Proteasen wie Matrix-Metalloproteinasen produzieren, die die Basalmembran, die das Endothel umgibt, zerstören können. M2-Makrophagen stimulieren auch die Angiogenese durch die Produktion von vaskulären endothelialen Wachstumsfaktoren.“
Also M2 Makrophagen verschlimmern die Tumorproblematik, die will man nicht in großen Mengen haben.“ (https://pubmed.ncbi.nlm.nih.gov/38583833/)
Nach der Aktivierung der Toll-like-Rezeptoren (TLRs) werden Makrophagen in großen Mengen aktiviert, was zu einem Makrophagen-Aktivierungssyndrom (MAS) führt, das die überschüssige Sekretion von Entzündungszytokinen anregt.
Die Entzündungsreaktion auf Spike-Protein besteht auch in einer Lymphopenie, die sich in einer Verringerung der CD4+ und CD8+ T-Zellen äußert.
Eine solche Beeinträchtigung der T-Zell-Antwort kann aus einer mangelhaften IFN-Produktion resultieren, die zu einer starken Erschöpfung der NK-Zellen führen kann.
Diese Ereignisse führen zu einem Ungleichgewicht in der angeborenen/erworbenen Immunantwort, zu einer verzögerten Virusabwehr und zu einer ungewöhnlichen Dominanz von hyperstimulierten Makrophagen und Neutrophilen in den verletzten Geweben.
Das Hämophagozytose-ähnliche Syndrom führt zu einer unwirksamen viralen Zytotoxizität und einer schwachen Antikörperproduktion.
Die unkontrollierte Verstärkung der Zytokinproduktion führt zu endothelialer Dysfunktion, ARDS, Gewebeschäden und Multiorganversagen, was der Ausgangspunkt für die Entwicklung der schweren und tödlichen Komplikationen der mod-RNA-Injektionen ist.
Anti-ACE2-Antikörper
Die Anti-ACE2-Antikörper können auch an ACE2 binden. Man hat es also mit einer Autoimmunreaktion gegen ACE2 zu tun. Die Symptomatik dürfte ähnlich sein.
“Anti-Idiotyp-Antikörper gegen ACE2 wurden bei Patienten nachgewiesen, die sich von einer COVID-19-Infektion erholt hatten, und es wurde die Hypothese aufgestellt, dass solche Antikörper nach einer SARS-CoV-2-Infektion oder -Impfung zu unerwünschten Ereignissen führen könnten, die eine Aktivierung des Immunsystems gegen ACE2 exprimierende Zellen, wie z. B. Neuronen, zur Folge haben. Unsere Daten zeigen klinisch überlappende Syndrome, die durch eine SARS-CoV-2-Infektion oder -Impfung ausgelöst werden und eine Positivität für Anti-ACE2-Antikörper aufweisen. Ihr Vorhandensein bei Fehlen anderer klassischer Autoimmunmarker für eine ZNS- oder PNS-Beteiligung deutet darauf hin, dass sie eine aktive Rolle im Zusammenhang mit einer abnormen Immunantwort spielen könnten.”
Anti AT1R-Antikörper
Nicht überraschend, eigentlich vorhersehbar, gibt auch Antikörper gegen AT1R. Das hatte ich bereits hier1 angeschnitten.
“Sechs dieser Antikörper (AT1R, ETAR, M2R, M3R, β2-adr-R, MASR) waren bei PACVS-Patienten signifikant (p < 0,0001) höher als bei den Kontrollpersonen nach der Impfung.” (Chronic Fatigue and Dysautonomia following COVID-19 Vaccination Is Distinguished from Normal Vaccination Response by Altered Blood Markers)
Verminderte Angiotensin-Rezeptor 1-Expression (AT1R) in den Hoden von ± AT1-Knockout-Mäusen führt zu männlicher Unfruchtbarkeit und GnRH-Reduktion (https://rbej.biomedcentral.com/articles/10.1186/s12958-021-00805-1)
Fazit
An dieser Stelle habe ich den Überblick über die Schadensstufe verloren. Wie viele Kaskaden wurden an diesem Punkt parallel losgetreten?
Gibt es etwas, was nicht dereguliert wurde?
An dieser Stelle gebe ich auf.
Könnt ihr nun verstehen, warum ich Pathways hasse?
Ich bräuchte nun ein virtuelles 3D Modell, in welches ich rein- und rauszoomen kann, um mich zu orientieren.
Das wäre mal eine Aufgabe für künstliche Intelligenz.
Freiwillige vor.
Update:
07.04.2025: Anti AT1R Antikörper
Chronic fatigue Syndrom - by DrBines verbales Vitriol

The human body is made of matter. When it comes to matter, quantity is important. The number of spikes that can be created by vaccination varies depending on the scholar, but it is estimated to be 100 to 10,000 times the number that would result from natural infection.
Quantitative analysis would be useful.
Der Link
https://phmpt.org/wp-content/uploads/2023/02/125742_S1_M4_4.2.1-vr-vtr-10741.pdf Seite 10
funktioniert nicht, auch nicht über archive.org