


Ugurs grenzdebile Schwachsinnsideen im Protein Design
Teil 3: Warum es eine grenzdebil schwachsinnige Idee war, Proline zu verbauen.

Im ersten Teil haben wir gelernt, warum es keine gute Idee ist, Codons zu „optimieren“.
Im zweiten Teil haben wir gelernt, warum man von dynamischen, großen Proteinen, deren Struktur und Funktionsmechanismus nicht kennt, die Finger lassen sollte.
Im dritten Teil geht es um das hochgelobte Prolin-Schloss, das angeblich die Struktur des Spike Proteins stabilisieren sollte, damit es, ja, was eigentlich?
Erneut werde ich die Fußnotenanzahl gering halten. Es war Pandemie! Panik! Da hat man nicht viel Zeit zu lesen. Ich werde mich also an die absolut notwendigsten Belege halten, die man an einem Wochenende locker und entspannt hätte lesen können.
Was war noch mal gleich das Prolin-Schloss? In Ugurs „Project Lightspeed“ findet sich dazu folgende Information in seinen Worten oder zumindest von ihm so genehmigt:
„Kurz bevor es an die Lungenzellen andockt, verändert sich das Spike-Protein von einer kompakten distelähnlichen Form in eine Struktur, die eher einem hohen, langgestreckten Kelch gleicht. Hat das Spike-Protein erst einmal an die Zelle angedockt, verändert es erneut seine Form, und ein Teil des Spikes wird ausgefahren wie eine Schnappmesserklinge, mit der es die Zellmembran durchsticht, sodass das Virus in die Zelle eindringen kann. Das Genom gelangt in die Zelle und kann dort vervielfältigt werden. Damit der Impfstoff wirken konnte, musste er also die Kelchform des Spike-Proteins nachbilden. Die Eingreiftruppen des Immunsystems würden so informiert, das Virus anzugreifen, bevor es zum Schnappmesser wurde – der Form also, die es nutzt, um in die Zelle einzudringen. Mit etwas Glück (Hervorhebung von mir) würde so der wirksame Andockprozess des Virus unterbrochen. […] Die Proteinstruktur, abgelöst vom Virus, war von Natur aus nicht formstabil (Hervorhebung von mir). Es war durchaus möglich, dass die körpereigenen Zellen, wenn die mRNA ihnen die Blaupause der Gensequenz für das Spike-Protein lieferte, eine leicht veränderte Struktur zusammenbaute und nicht die exakte Form (S. 103).“ […] „2012 entwickelte [Barney Graham] einen Ansatz, spike-ähnliche Antigene in der Form, die sie vor Eintritt in die Zelle haben, zu bewahren. Das gab Hoffnung, endlich ein sicheres RSV-Vakzin zu finden. (S. 104)“
Hier zitiert der Autor einen Beleg, der nicht zum Text passt. RSV ist kein Coronavirus. Was RSV mit dem Coronavirus zu tun hat, das erklärt er praktischerweise auch nicht.
In diesem Zitat beschreibt der Autor die spekulative Funktionsweise des Prolin-Schlosses[1] [2](prolin lock), wie es von BioNTech/Pfizer geplant war. Man wollte das Spike-Protein ein einer Prä-Fusions Konformation gefangen halten, damit es nicht in die Zelle eindringt.
Das hat übrigens nicht funktioniert. Zwei Proline sind generell instabil und man hat das Spike-Protein aktiv und frei zirkulierend im Blut nachgewiesen[3]. Um die Form zu stabilisieren braucht es wohl 6 Proline[4].
Diese Proline (übrigens essentielle Aminosäuren, die man über die Nahrung zu sich nehmen muss) ziehen aber Probleme nach sich, die hier verschwiegen werden. Der Austausch von 2 Aminosäuren kann die Struktur eines Proteins auf unvorhergesehene Weise verziehen und verändern über sogenannte „Einflüsse über lange Distanz“. Das habe ich in meiner Doktorarbeit selbst erleben dürfen. Zwei Mutationen (I84V und S87R) reichten, meine tote Katze (CAT I) wiederzubeleben.
Ugur weiß das auch, er schreibt es ja selbst:
Die Proteinstruktur, abgelöst vom Virus, war von Natur aus nicht formstabil. Es war durchaus möglich, dass die körpereigenen Zellen, wenn die mRNA ihnen die Blaupause der Gensequenz für das Spike-Protein lieferte, eine leicht veränderte Struktur zusammenbaute und nicht die exakte Form (S. 103).“
Wenn man es genau nimmt, schreibt Ugur da sogar, dass anhand seiner mRNA ein anderes Protein herauskommt, als das natürliche Spike. Was diese Unterschiede für Folgen nach sich ziehen könnten hat er aber nicht auf dem Schirm. Er scheint nicht einmal zu ahnen, dass es ein Problem sein könnte, wenn ein Protein nicht formstabil ist. Er ahnt nichts, von den zugrundeliegenden dynamischen Gleichgewichten bzw. er ist nicht reflektiert genug, an dieser Stelle kurz innezuhalten und nachzudenken, was er da gerade geschrieben hat.
Woher kam die schwachsinnige Idee zum Prolin-Schloss eigentlich ursprünglich?
Dieses Prolin-Schloss ist vom NIH (National Institute for Health) Patentiert unter Patentnummer US10960070B2[5]. Patenthalter ist unter anderem der in Ugurs Buch erwähnte Barney Graham.
Es wurden basierend auf diesem Patent zwei Aminosäuren gegen Proline ausgetauscht K986P und V987P[6].
Graham arbeitet für das NIH, das National Institute for Health, Faucis Königreich in den USA. Moderna hat für die Verwendung des Prolin Schlosses von Graham Lizenzgebühren gezahlt bzw. zumindest einen nicht-exklusiven Lizenzvertrag abgeschlossen[7] und das für ein nicht funktionierendes Produkt. Inwieweit BioNTech/Pfizer ebenfalls möglicherweise Lizenzgebühren gezahlt haben ist unbekannt. Es wird aber offensichtlich, dass bei der Verwendung des Prolin-Schlosses ein möglicher finanzieller Vorteil für Faucis US-Behörde entstanden sein könnte. Die Behörde, die mit drakonischen Vorschriften dann die US Behörde in die Spritze getrieben hat.
Leider wurden von Seiten BioNTech/Pfizer keine Daten vorgelegt, ob das Prolin-Schloss überhaupt funktioniert, bevor es zusätzlich in einem „rational design“ Ansatz verbaut wurde.
Den Unterschied zwischen rational design und directed evolution habe ich bereits in einem früheren Substack ausführlich erklärt.
Um abzuschätzen, wie „sicher“ rationales Design in Proteinen ist, gibt es einen schönen Vergleich der von Englander[8] in seinem Paper illustriert, wie weit unser „Verständnis“ von Proteinfaltung ist:
„So kann man beispielsweise den Weg einer Multitonnen-Rakete durch 150 Millionen Meilen freien Raum bis zu einer punktgenauen Landung auf dem Mars berechnen. Die Gleichungen, die die Raumfahrt bestimmen, sind genau bekannt, die Computerleistung ist ausreichend, und die zu kontrollierende Strecke ist klar. Die Berechnung der strukturellen Reise winziger Proteinmoleküle durch die Submikrometer des Faltungsraums hat sich als schwieriger erwiesen.“ – Die erste Marslandung scheiterte an einem Rechenfehler…[9]
Das wäre mittels einer Biacore[10] Messung sehr einfach gewesen. Physikalisch nennt man eine Biacoremessung (benannt, nach der Firma, die das entwickelt hat, damals in den 1990er Jahren[11]) auch Oberflächenplasmonenresonanzspektroskopie. In Teil 5 werde ich darauf eingehen, welche Messungen ich durchgeführt hätte. Es ist in diesem Zusammenhang nicht wichtig, zu verstehen, wie Biocore funktioniert. Es reicht, zu verstehen, dass es eine über 20 Jahre etablierte Messmethode gegeben hätte, mit der man hätte überprüfen können, ob das modifizierte Spike bindet, wie stark es bindet, an welche Rezeptoren es sonst so bindet und ob man das modifizierte Protein wieder vom Rezeptor herunter bekommt, wenn es gebunden hat. Ja wenn man gewusst hätte, dass es eine solche Messmethode gibt und sie zum Einsatz gekommen wäre.
Es gibt aus den Pfizer eigenen Unterlagen aber durchaus vergleichbare Datensätze, die zwar ungenauer sind, aber eine Bilderbuch Bindungskurve des eigentlich nicht mehr bindungsfähigen Spike an ACE2 zeigen[12]:
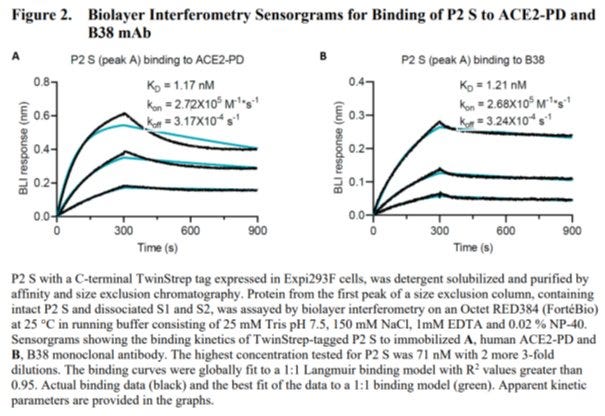
Eigentlich soll das Schaubild zeigen, dass der vom Immunsystem nach der Impfung produzierte Antikörper an das Spike-Protein bindet (rechtes Bild). ABER ein in einer Präfusionskonfirmation geblocktes Spike-Protein hätte nicht an ACE2 Binden dürfen (linkes Bild) sondern ein flache Linie oder sehr niedriges Signal ergeben sollen. Man wusste also zumindest aus diesen Messwerten, dass das Prolin-Schloss nicht funktionierte, an ACE2 bindet und dann nicht mehr herunterzuspülen ist (sonst wäre das Signal gegen Ende wieder in Richtung Nulllinie abgefallen) und somit den Rezeptor blockiert. Welche Folgen eine solche dauerhafte Blockade nach sich ziehen würde hat man sich wohl nicht gefragt.
Letztendlich ist es aber egal, ob das Prolin-Schloss die gewünschte Funktion ausübt oder nicht, weil allein die Idee ein Prolin-Schloss zu verbauen, egal, ob es funktioniert oder nicht, im Desaster enden musste und zwar aus altbekannten Gründen, die ich nun erläutern werde.
Generell sind Proline in der Proteinfaltungsliteratur als, milde ausgedrückt, problematische Aminosäuren bekannt[13].
„Ein auffälliges, wiederkehrendes Merkmal des intermolekularen Aufbaus, das sowohl zum Domänenaustausch als auch zur Aggregation führt, sind Prolinreste. Es scheint, dass die Prolin-Isomerisierung der Schalter ist, der die Umwandlung zwischen monomeren und oligomeren Formen auslöst.“[14]
Das heißt im Klartext, Proline können dazu führen, dass Proteine Amyloide bilden. Amyloide sind krank machende Proteinaggregate, die man als Krankheitsbild in der Medizin als Amyloidose[15] kennt. Unter durch Proteinfehlfaltung verursachte Krankheiten fallen des Weiteren Alzheimer, Parkinson, Spongiforme Encephalitis, Diabetes Typ II. Die Menge des fehlgefalteten Proteins kann in einigen Organen durchaus den Kilogrammbereich erreichen[16].
Ein erfahrener Protein Engineer wird daher Proline meiden wie der Teufel das Weihwasser.
Die Natur weiß auch, dass Proline ein Problem darstellen, daher dienen Proline für Faltungshelferproteine (Chaperone) häufig als Erkennungssignal. Das wird sicherlich seinen Grund haben.
Als wäre das nicht schon genug Warnung, AUF KEINEN FALL PROLINE ZU VERBAUEN, ist aus der Literatur bekannt, dass der Einbau eines Prolins in das Hepatitis Spike Protein dieses neurotoxischer macht[17].
Und selbst wenn all das oben genannte kein Problem gewesen wäre und alles so geklappt hätte, wie man sich das ausgedacht hätte, wäre es trotzdem schief gegangen, weil man eine Kleinigkeit nicht bedacht hatte: eine Regel, die sich „Leben auf Messers Schneide der Löslichkeit (Life on the edge (of solubility)) nennt.
„Die Aufrechterhaltung des richtigen Gleichgewichts in den Populationen der verschiedenen Zustände von Proteinen ist von großer Bedeutung, da selbst geringfügige Veränderungen in diesen Populationen langfristig zu Krankheiten führen können.“[18]
Ich habe einen derartigen Gleichgewichtseffekt in meiner Promotion tatsächlich selbst erlebt, damals aber nicht verstanden, was es damit auf sich hatte. Das wurde mir tatsächlich erst klar, als ich mich für diese Reihe vertieft noch einmal in die aktuelle Literatur eingelesen habe.
Ich hatte in m einer Promotion[19] ein seltsames Phänomen, wenn ich mein Protein nach Größe sortiert noch einmal mit einer Sephadex 200 Säule gereinigt habe. Mein trimeres Protein ergab 3 Peaks, obwohl es bereits sauber war. Ich wollte es über diese Säule eigentlich nur umpuffern.
Wenn ich einen dieser drei Peaks noch einmal über die Säule schickte, spaltete sich das Protein erneut in diese drei Peaks auf: Trimer, Hexamer, Nonamer. Es gab also, aus einem mir unbekannten Grund, ein dynamisches, stabiles, sich immer wieder einstellendes Gleichgewicht zwischen diesen drei Formen.
Wenn also das Prolin-Schloss theoretisch wirklich funktioniert hätte, wäre dadurch das sich immer wieder einstellende Gleichgewicht zwischen den diversen Konformationen des Spike Proteins ohnehin durcheinandergekommen und hätte zu einem Problem geführt. Möglicherweise wären dann die Prä-Fusions-Proteine aggregiert, weil es von ihnen zu viele gegeben hätte im Verhältnis zu den anderen Konformationen?
Vielleicht ist die Antwort aber auch deutlich einfacher und die gleiche wie in meiner Promotion:
Das Spike zeigt anscheinend ein ähnliches Verhalten wie mein trimeres Protein aus meiner Doktorarbeit:
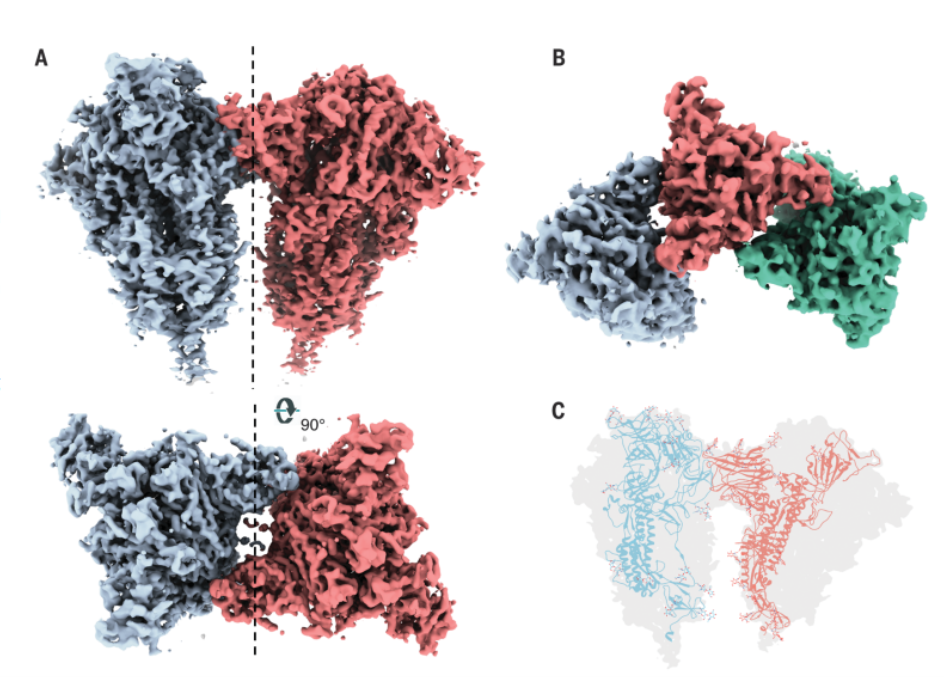
Es bildet Trimere, Hexamere und Nonamere [21], was man meines Wissens nicht ansatzweise in Betracht gezogen hat. Welchen Einfluss diese Verhalten auf die Immunreaktion haben könnte, auch bei hybriden Spike Proteinen bei Kreuzschlumpfung, kann ich nicht ansatzweise abschätzen, zumal das Spike-Protein auch noch an Polysorbat 80 bindet und dann Virusähnliche Partikel bildet. DAS hat aber nichts mit den Prolinen zu tun aber mit dynamischen Gleichgewichten, denn möglicherweise stehen auch in diesem Fall die Trimere, Hexamere und Nonamere in Lösung in einem bestimmten, fixten Gleichgewicht? Vielleicht ist es aber auch nur eine normale Wahrscheinlichkeitsverteilung, dass Trimere am wahrscheinlichsten sind, dann Hexamere und dann Nonamere und das einfach an der Konzentration der Proteine in Lösung liegt.
Novavax war nie eine Alternative

Novavax ist auf den ersten Blick ein relativ klassischer Protein-Impfstoff mit Adjuvans (Wirkverstärker). Novavax besteht also aus dem modifizierten Spike Protein, welche sich zu virusartigen Partikeln zusammengelagert haben und mit einem Adjuvans kombiniert werden. Die Tücken stecken im Detail.
Erschwerend kommt zu alldem noch hinzu, dass man durch die in Teil 1 erwähnte Codonoptimierung, die Proteinmenge erhöhen und so optimieren wollte, weil man wohl dachte „viel hilft viel“.
Was man dabei nicht bedacht hat ist, dass Proteine in hohen Konzentrationen thermodynamisch instabil sind und daher zur Aggregation neigen. Es gibt daher einen evolutionären Druck, die Konzentration eines Proteins so zu optimieren, dass es löslich bleibt und eine gewisse Konzentration nicht überschreitet. Je niedriger die Konzentration, desto stabiler. Proteine sind daher von Natur aus genau so stabil, dass sie in den Konzentrationen, in welcher sie benötigt werden, vorliegen[20]. Stabilität und Konzentration hängen somit voneinander ab und beeinflussen sich möglicherweise.
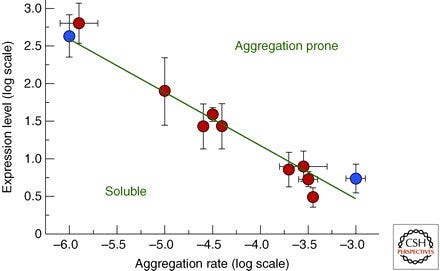
Das Bild oben zeigt die Korrelation zwischen In-vivo-mRNA-Expressionsniveaus und In-vitro-Proteinaggregationsraten für eine Reihe von 11 menschlichen Proteinen, die entweder mit Krankheiten in Verbindung (rot) oder nicht in Verbindung (blau) stehen.
Hier hätten wir auch die Verbindung zwischen mRNA-Ablesegeschwindigkeit und Ablesemenge bzw. der potentiellen Langlebigkeit der modRNA zur Proteinkonzentration und den damit potentiell einhergehenden Löslichkeitsproblemen. Und das zusätzlich zur Stabilitätsveränderung durch das Prolin-Schloss.
Vendruscolo M sagt in seiner Publikation sehr klar und eindeutig:
Es wurde „gezeigt, dass die Grenze der "sicheren" Konzentration von Proteinen in lebenden Systemen erreicht ist, wenn die Stabilität des alternativen Aggregatzustands mit der des nativen Zustands vergleichbar ist. Es ist daher von großer Bedeutung, die gut etablierte Charakterisierung der Struktur, Faltung und Stabilität nativer Zustände durch eine ähnliche Analyse der Struktur, des Aufbaus und der Stabilität anderer Zustände zu ergänzen - von ungefalteten und teilweise gefalteten Spezies, einschließlich nativ ungefalteter Zustände, bis hin zu aggregierten Spezies wie Amyloidfibrillen.“
Wurden diese Charakterisierungen des Prä-Fusions-Spikes im Vergleich zum natürlichen Spike Protein durchgeführt?
Wurden überhaupt die Verhältnisse der verschiedenen Spike-Konformationen in Lösung bestimmt?
Wurden diese Charakterisierungen des Spikes mit den zwei zusätzlichen Prolinen und ohne Proline, also im natürlichen Zustand, im Vergleich durchgeführt?
War man sich des Problems überhaupt bewusst?!
Um das Problem zu erfassen, hätte es gereicht, vier einfach verständliche, 10 bis 20 Jahre alte, Übersichtsartikel zu lesen:
Englander SW, Mayne L. The nature of protein folding pathways. Proc Natl Acad Sci U S A. 2014 Nov 11;111(45):15873-80. doi: 10.1073/pnas.1411798111. Epub 2014 Oct 17. PMID: 25326421; PMCID: PMC4234557. https://pubmed.ncbi.nlm.nih.gov/25326421/
Tsytlonok, M., & Itzhaki, L. S. (2013). The how’s and why’s of protein folding intermediates. Archives of Biochemistry and Biophysics, 531(1–2), 14–23. https://doi.org/10.1016/j.abb.2012.10.006
Dobson CM. Protein folding and misfolding. Nature. 2003 Dec 18;426(6968):884-90. doi: 10.1038/nature02261. PMID: 14685248. https://pubmed.ncbi.nlm.nih.gov/14685248/
Vendruscolo M, Knowles TP, Dobson CM. Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harb Perspect Biol. 2011 Dec 1;3(12):a010454. doi: 10.1101/cshperspect.a010454. PMID: 21825020; PMCID: PMC3225949. https://pubmed.ncbi.nlm.nih.gov/21825020/
[1] Cross, R. (2020, September 29). https://cen.acs.org/pharmaceuticals/vaccines/tiny-tweak-behind-COVID-19/98/i38. C&En. Retrieved February 19, 2023, from https://cen.acs.org/pharmaceuticals/vaccines/tiny-tweak-behind-COVID-19/98/i38
[2] Pallesen J, Wang N, Corbett KS, Wrapp D, Kirchdoerfer RN, Turner HL, Cottrell CA, Becker MM, Wang L, Shi W, Kong WP, Andres EL, Kettenbach AN, Denison MR, Chappell JD, Graham BS, Ward AB, McLellan JS. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A. 2017 Aug 29;114(35):E7348-E7357. doi: 10.1073/pnas.1707304114. Epub 2017 Aug 14. PMID: 28807998; PMCID: PMC5584442.
[3] Ogata AF, Cheng CA, Desjardins M, Senussi Y, Sherman AC, Powell M, Novack L, Von S, Li X, Baden LR, Walt DR. Circulating Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Vaccine Antigen Detected in the Plasma of mRNA-1273 Vaccine Recipients. Clin Infect Dis. 2022 Mar 1;74(4):715-718. doi: 10.1093/cid/ciab465. PMID: 34015087; PMCID: PMC8241425.
[4] Lu M, Chamblee M, Zhang Y, Ye C, Dravid P, Park JG, Mahesh KC, Trivedi S, Murthy S, Sharma H, Cassady C, Chaiwatpongsakorn S, Liang X, Yount JS, Boyaka PN, Peeples ME, Martinez-Sobrido L, Kapoor A, Li J. SARS-CoV-2 prefusion spike protein stabilized by six rather than two prolines is more potent for inducing antibodies that neutralize viral variants of concern. Proc Natl Acad Sci U S A. 2022 Aug 30;119(35):e2110105119. doi: 10.1073/pnas.2110105119. Epub 2022 Aug 22. PMID: 35994646; PMCID: PMC9436349.
[5] Graham, B. (2016, October 25). US10960070B2 - Prefusion coronavirus spike proteins and their use - Google Patents. https://patents.google.com/patent/US10960070B2/en
[6] Hildt, E. (2022). Übersicht über die in der EU zugelassenen COVID-19-Impfstoffe – von der Technologie über die klinische Prüfung zur Zulassung. Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz, 65(12), 1237–1243. https://doi.org/10.1007/s00103-022-03600-4 https://link.springer.com/article/10.1007/s00103-022-03600-4
[7] https://d18rn0p25nwr6d.cloudfront.net/CIK-0001682852/72a717be-931e-4d92-bd4e-e7ad4652a97d.pdf
[8] Englander SW, Mayne L. The nature of protein folding pathways. Proc Natl Acad Sci U S A. 2014 Nov 11;111(45):15873-80. doi: 10.1073/pnas.1411798111. Epub 2014 Oct 17. PMID: 25326421; PMCID: PMC4234557. https://pubmed.ncbi.nlm.nih.gov/25326421/
[9] Spiegel, D. (1999, October 1). a-e4547083-0001-0001-0000-000000044777. DER SPIEGEL, Hamburg, Germany. https://www.spiegel.de/wissenschaft/mensch/mars-climate-orbiter-absturz-wegen-leichtsinnsfehler-beim-rechnen-a-44777.html
[10] Cytiva. (n.d.). Biacore. https://www.cytivalifesciences.com/en/us/about-us/our-brands/biacore
[11] Wikipedia contributors. (2021, May 10). Biacore. Wikipedia. https://en.wikipedia.org/wiki/Biacore
[12] https://phmpt.org/wp-content/uploads/2023/02/125742_S1_M4_4.2.1-vr-vtr-10741.pdf Seite 10
[13] Wedemeyer WJ, Welker E, Scheraga HA. Proline cis-trans isomerization and protein folding. Biochemistry. 2002 Dec 17;41(50):14637-44. doi: 10.1021/bi020574b. PMID: 12475212. https://pubmed.ncbi.nlm.nih.gov/12475212/
[14] Tsytlonok, M., & Itzhaki, L. S. (2013). The how’s and why’s of protein folding intermediates. Archives of Biochemistry and Biophysics, 531(1–2), 14–23. https://doi.org/10.1016/j.abb.2012.10.006
[15] Wikipedia-Autoren. (2004d). Amyloidose. de.wikipedia.org. https://de.wikipedia.org/wiki/Amyloidose
[16] Dobson CM. Protein folding and misfolding. Nature. 2003 Dec 18;426(6968):884-90. doi: 10.1038/nature02261. PMID: 14685248. https://pubmed.ncbi.nlm.nih.gov/14685248/
[17]Singh M, Kishore A, Maity D, Sunanda P, Krishnarjuna B, Vappala S, Raghothama S, Kenyon LC, Pal D, Das Sarma J. A proline insertion-deletion in the spike glycoprotein fusion peptide of mouse hepatitis virus strongly alters neuropathology. J Biol Chem. 2019 May 17;294(20):8064-8087. doi: 10.1074/jbc.RA118.004418. Epub 2019 Mar 1. PMID: 30824541; PMCID: PMC6527167. https://pubmed.ncbi.nlm.nih.gov/30824541/
[18] Vendruscolo M, Knowles TP, Dobson CM. Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harb Perspect Biol. 2011 Dec 1;3(12):a010454. doi: 10.1101/cshperspect.a010454. PMID: 21825020; PMCID: PMC3225949. https://pubmed.ncbi.nlm.nih.gov/21825020/
[19] FreiDok plus - Nucleotide exchange and excision technology (NExT) DNA shuffling And Mutational studies on chloramphenicol actetyltransferase I (CATI). (n.d.). https://freidok.uni-freiburg.de/data/11610
[20] Vendruscolo M, Knowles TP, Dobson CM. Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harb Perspect Biol. 2011 Dec 1;3(12):a010454. doi: 10.1101/cshperspect.a010454. PMID: 21825020; PMCID: PMC3225949. https://pubmed.ncbi.nlm.nih.gov/21825020/
[21] Bangaru S, Ozorowski G, Turner HL, Antanasijevic A, Huang D, Wang X, Torres JL, Diedrich JK, Tian JH, Portnoff AD, Patel N, Massare MJ, Yates JR 3rd, Nemazee D, Paulson JC, Glenn G, Smith G, Ward AB. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science. 2020 Nov 27;370(6520):1089-1094. doi: 10.1126/science.abe1502. Epub 2020 Oct 20. PMID: 33082295; PMCID: PMC7857404. https://pubmed.ncbi.nlm.nih.gov/33082295/
Wunderbarer Artikel wieder, großes Dankeschön!
Ein weiterer Gedanke noch dazu obendrauf:
Ist es nicht auch zusätzlich sinnlos, das Prolin-Schloss in der S2 Untereinheit einzubauen, wo doch die S1 Untereinheit nachgewiesenermaßen im Blut frei herumschwimmt?
Das erklärt wunderbar Florian Schilling in einem Video:
https://www.florianschillingscience.org/post/good-spike-bad-spike
Welche Konformationen kann diese Untereinheit dann ohne die ohnehin erfolglose Fixierung durch das Prolin-Schloss annehmen? --> Bestimmt auch super gut untersucht worden (Ironie).
Viele Grüße
U.Cl.