
Directed (Protein) Evolution
Das Spike stinkt mir nach gene shuffling und phage display

Dieser Artikel war nicht geplant. Ich bin zwar an einem Artikel über die Anfängerfehler im Protein Engineering, die beim Spike passiert sind (und das sind echt viele, da wird jemandem vom Fach kotzübel, ich muss da aber noch ein wenig aktuellere Literatur lesen, das schüttel ich nicht so aus dem Ärmel wie diesen Artikel). Das Thema directed Evolution wollte ich darin aber nicht anschneiden. Ich habe mir zwar seit zwei Jahren den Mund fusselig geredet, dass das Spike mir nach directed evolution stinkt, aber nicht mal die Doctors for Covid Ethics haben den Hinweis verstanden, obwohl ich ihnen im Chat meine alten Buchkapitel, Paper und meine Dissertation verlinkt habe.
Caveat: Jeder Wissenschaftler sieht das Spike NATÜRLICH durch den Rahmen/Frame seiner eigenen wissenschaftlichen Laborerfahrungen. Für mich sieht so was daher natürlicherweise nach directed Evolution aus, weil ich das gemacht habe, und andere Methoden, wie man das hätte machen können, möglicherweise gar nicht kenne. Das heißt NICHT, dass das Spike wirklich so gebaut wurde, es wäre aber eine theoretische, sehr einfache Möglichkeit, die jeder Praktikant soweit unbemerkt durchziehen könnte.
Da aufgrund des versteckten Interviews von Project Veritas mit Jordon Trishton Walker[i], der angeblich ein Forscher bei Pfizer in NY ist (ich sage angeblich, weil es unter den Wissenschaftlern gerade eine heiße Diskussion gibt, ob er wirklich so eine Position innehaben kann, sein Lebenslauf würde so eine Position nicht hergeben…) das Thema directed Evolution gerade hochkocht, es aber gerade mal eine Handvoll Wissenschaftler geben dürfte, die auf dem Gebiet gearbeitet haben und die aus meinem damaligen Labor arbeiten nun für BAYER, GSK oder BioNTech (Jaaaaaaa, meine experimentelle andere Hälfte, die ein praktisch identisches Projekt, aber mit anderen Eiweiß (Er: Monomer TEM-1, Ich: Trimer CAT 1) arbeitet nun bei BioNTech, nur so, ob die dazu in er Lage wären: JA), bleibt der Artikel halt an mir hängen, weil es in der Wissenschaftlergemeinde der Widerstandes international außer mir keinen zu geben scheint, der auf dem Gebiet unterwegs war. Irgendwas habe ich wohl falsch gemacht. Meine Mitdoktoranden verdienen mit ihrem Wissen nun ordentlich Schotter und ich schreibe kostenlose Substacks.
Wissenschaftler sind nicht mehr Ziel meiner Artikel (das habe ich ehrlich gesagt aufgegeben), sondern die wachen, interessierten Normalos ohne Studium aber mit ordentlicher Ausbildung. Daher, liebe Wissenschaftler, wenn ihr euch doch auf meinen Substack verirrt, ich verlinke die hardcore Artikel, der Text bleibt aber hoffentlich auf einem allgemeinverständlichen Niveau (obwohl ich laut meiner Betreuerin im Referendariat erhebliche Defizite in der didaktischen Reduktion habe. Mir wurde zumindest bescheinigt, dass ich am besten in die Erwachsenenbildung passe und da bin ich nun wohl auch gelandet mit meinem Substack). Ich versuche auf ein VHS Niveau runterzubrechen, kann aber nicht garantieren, dass das auch funktioniert. Wissenschaftlich wird dabei sicherlich Einiges auf der Strecke bleiben.
Da ich keine Lust mehr habe es allen, die mich auf Telegram anschreiben, einzeln zu erklären, schreibe ich nun diesen Artikel, der nicht durch Evas (danke liebe Eva dafür) Rechtschreibkontrolle gehen wird (Kapitel 1 zu Project Light Speed ist tatsächlich schon in der Rechtschreibkontrolle). Lebt mit den Tippfehlern, der Text soll schnell raus, wird spontan aus dem Handgelenk geschüttelt und da müsst ihr halt mit Tippfehlern leben. Auf den Inhalt kommt es an, nicht auf die Schönheit. Aus einem schönen Teller wird man nicht satt, sagte immer meine Oma. Dieser Text ist unkontrollierte, ungebremste 100% DrBine.
Ironie und Sarkasmus wird es in diesem Artikel wohl auch wenig geben, das macht sprachlich einiges an Mühe und geht nur bei echt schlechten Büchern und richtig dummen Aktionen. Hier bleibt es also sachlicher und wird nicht ganz so witzig. Ich werde aber versuchen ironische Seitenhiebe spontan einzubauen, versprochen.
Was bedeutet directed Evolution eigentlich?
Ich würde directed Evolution am ehesten als durch die Umweltbedingungen zielgesteuerte Evolution bezeichnen. Also eigentlich das, war in der Natur auch passiert. Dieser evolutionäre Druck durch die Umwelt wird im Labor durch verschiedene Methoden in vitro (im Reagenzglas bzw. irgendwelchen anderen Gas-oder Plastikwaren (das ist ein leidiges Kapitel für sich)) simuliert.
Wie funktioniert directed Evolution im Labor?
Da ich immer noch genauso stinkfaul wie als Doktorand bin, werde ich mich bei meinen alten Bildern und Zeichnungen bedienen und sie nicht ins Deutsche übersetzen, sondern einfach mit Bildschirmfotos aus den alten Artikeln holen. Hab ich schon in meiner Doktorarbeit so gemacht, ich war zu faul die Fundustexte vom Laborserver ins Englische zu übersetzen, daher ist meine Doktorarbeit eine Mischung aus Deutsch und Englisch. Gab zwar 0,5 Notenabzug, war mir aber wurscht, das war meine Lebenszeit mir wert.
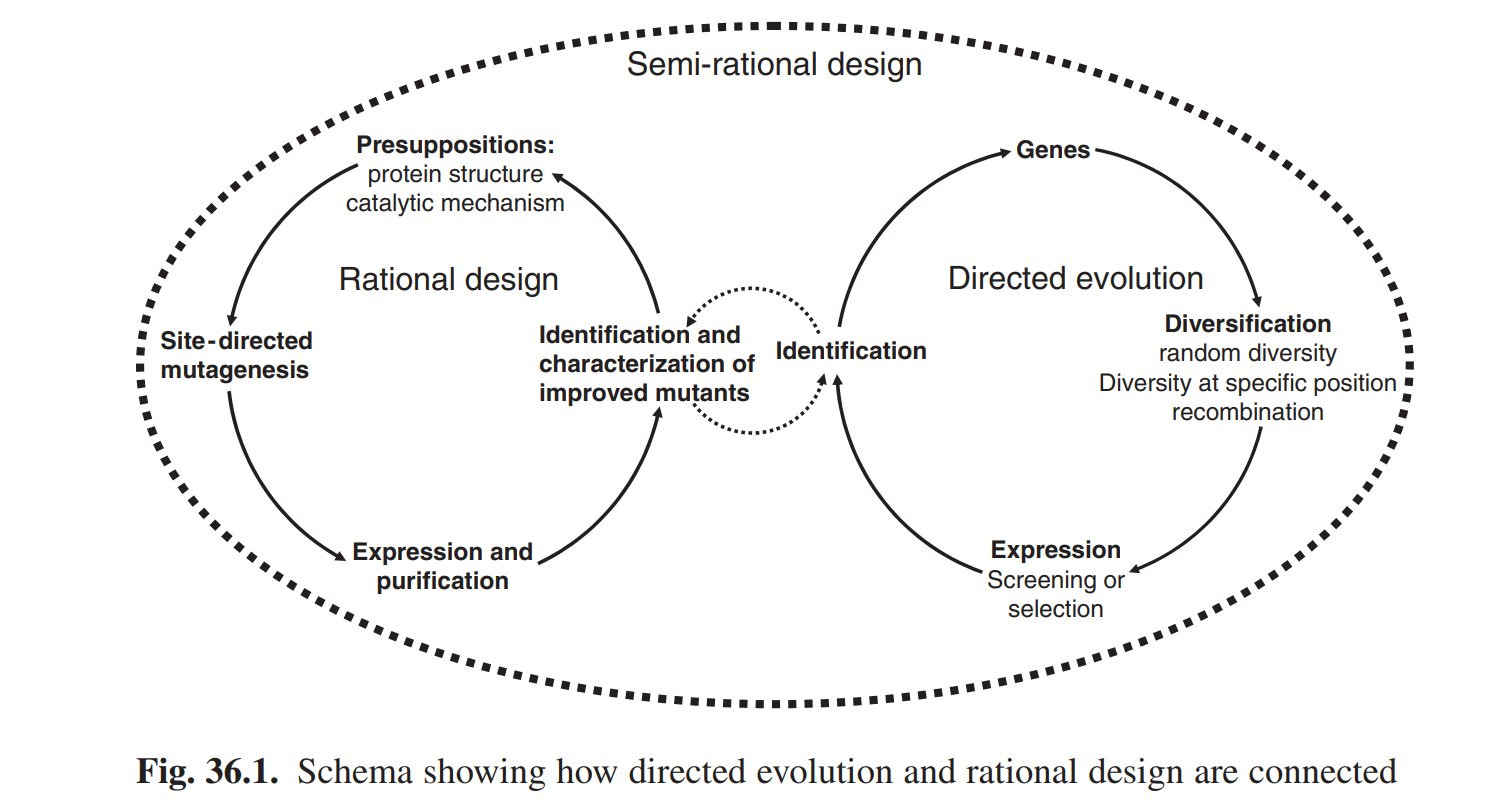
Das Schaubild stammt aus einem meiner alten Buchkapitel[i] und ist eine Übersicht, wie directed Evolution im Labor grundsätzlich funktioniert. Achtung: Das ist mein Kenntnisstand vor 15 Jahren. Ich habe das Thema seither nicht mehr verfolgt, weil Menschen wie ich wegen des Wissenschaftszeitvertragsgesetzes[ii] an Unis persona non grata, also nicht mehr einstellbar sind, weil es nur befristete Stellen gibt und man sich mit anstrengenden Selbstdenkern nicht langfristig (finanziell und nervlich) belasten will.
Es gibt zum einen die Möglichkeit Proteine (Eiweiße) auf einer rationalen Basis, also am Computer, zu mutieren und dann sein rationales Design auszuprobieren. Würde ich nicht empfehlen. Proteinfaltung und Modellierung am Computer waren damals noch in den Kinderschuhen, man versteht Proteinfaltung auch heute nicht ansatzweise und die Effekte von einzelnen Aminosäurenaustauschen (Aminosäuren sind die Bausteine der Proteine), kann man weder vorhersagen noch erklären (außer mit sehr viel heißer Luft, die schön klingt aber nichts sagt, wie bei Politikern. Die meisten Paper über Proteinfaltung sind heiße Luft, is aber meine persönliche Meinung).
Wer es eilig hat, wählt den Zweig der directed Evolution.
Dierectes Evolution hat mehrere Vorteile, die man nicht unterschätzen sollte:
1. Man muss die Struktur seines Proteins nicht kennen
2. Man muss die Funktionsweise seines Proteins weder kennen noch verstehen
3. Man muss keine Ahnung von Proteinfaltung haben.
4. Man muss sich keinen Kopf über den Effekt der Mutationen machen, was nicht funktioniert, fliegt im Screening einfach raus.
Kann also jeder Praktikant unauffällig nebenbei machen, sobald ein System sauber und robust aufgesetzt ist und die experimentellen Unwägbarkeiten behoben sind. Selbst in gemeinsam genutzten Laboren würde dem Wissenschaftler an der Nachbarbench nicht unbedingt auffallen, was der Kollege da eigentlich gerade treibt.
=== Schritt 1: Ich mutiere mir mein Gen ===
Als erstes muss man sein Gen diversifizieren. Wie haben ja in den letzten 3 Jahren gelernt, wie wichtig Diversität für die Gesellschaft ist. Je diverser die Genbibliothek, desto besser (also mehr als nur m/w/d + 20 weitere Geisteskrankheiten). Wir bewegen uns hier in Bereichen von mathematischen Potenzen, also seeeeeeeeeeeeeeeeeehr großen Zahlen mit vielen, vielen Nullen. Kurz gesagt, SEHR GROSS und SEHR VIEL also SEHR DIVERS (aber ohne blaue Haare und Selbstverstümmelung).
Wie bekomme ich mein Gen divers?
Mittels der sogenannten ErrorProne PCR[iii]. PCR kennen wir alle mittlerweile leider aus leidvoller dreijähriger Erfahrung, darauf wie die funktioniert gehe ich daher nicht mehr ein. Im Fall der directed Evolution nimmt man keinen Rachenabstrich, sondern 50ng (also sehr wenig) von seinem Gen, das man diversifizieren will. Im Prinzip ist eine ErrorProne PCR eine normale PCR bei der man die nervige Fehlerrate der ohnehin chronisch unzuverlässigen Taq Polymerase (die kennen wir seit 3 Jahren auch zur Genüge) noch einmal durch die Zugabe von Mangan (in Ionenform Mn2+, nicht als Metall) weiter hochtreibt. Man kann noch Magnesium (Mg2+) dazu tun, habe ich persönlich aber nicht gemacht, ich habe nur Mn2+ verwendet.
Kleine Anmerkung: Einige der Puffer der Corona PCR-Tests enthielten Mangan, z. Bsp. der von Roche…. Ein Schelm wer Böses dabei denkt.
Taq macht also bei der Produktion des Gens ordentlich Fehler, sogenannte Punktmutationen (und noch andere wie frameshift und dergleichen). Da Taq auch keine Korrekturlesefunktion hat wie ihr/e Kolleg*Innen Vent und Pfu (Ich habe keine Ahnung, ob die im Deutschen nun männlich oder weiblich sind. Die Vent, der Pfu oder umgekehrt), fällt das Taq nicht auf, sie macht einfach weiter und setzt Fehler auf Fehler.
=== Schritt 2: Deutschland sucht die perfekte Mutante/den perfekten Mutant/die perfekten Mutanten===
Dieses nun diversifizierte Gen baut man sich in einen Organismus, mit dem man eine darwinsche Selektion (also es überleben nur die Stärksten oder Passenden) durchführt. Je nach dem was und wie man selektieren will kann man in Bakterien arbeiten (hab ich gemacht) und einfach schauen, ob sie unter den Selektionsbedingungen überleben oder man sucht nach Protein:Protein Interaktion, also schaut, ob ein Eiweiß an ein anderes Eiweiß bindet. Da die meisten Leser sich wohl dafür interessieren, wie man das Spike hätte mit diesen Methoden mutieren können, gehen wir die Schritte für die Selektion der Protein:Protein Interaktion durch.
Hier würde ich als Organismus einen Bakteriophagen wählen. Also ein Bakterienvirus. Da gibt es Einige zur Auswahl[iv]. Findet man schön ausführlich bei Wikipedia erklärt, wie das funktioniert und bei wissenschaftlichen Artikeln ist Wikipedia teils noch echt OK. Diese Art des Screenings nennt sich Phage Display[v]. Auch das erklärt Wikipedia sehr schön, da brauche ich mich nicht widerholen, sonst wird es hier zu lang.
Man tauscht die „Füßchen“ eines Phagen, gegen das Protein der Wahl aus, das man auf seine Bindungseigenschaften untersuchen möchte und dann lässt man den Phagen mit seinen neuen „Füßchen“ binden. Der Auswahl des Zieles, an die der Phage binden soll, sind keine Grenzen gesetzt (außer man hat das andere Protein halt hat nicht. Nicht alles kann man direkt aus dem Katalog bestellen). Im Falle des Spike Proteins würde man schauen, ob die mutierten Spike-Füßchen des Phagen an ACE2 binden. Man kann verschiedene Varianten von ACE2 für verschiedenen Ethnien ausprobieren. Dem Screening Röhrchen ist wurscht, mit welcher Variante man es beschichtet.
Anmerkung: Das klingt jetzt alles einfacher, als es experimentell wirklich ist. Die Füßchen müssen schon zum Phagen passen, sind sie zu groß, funktioniert es nicht, man muss sich schon das richtige Phagenhaustier aussuchen für seine Experimente, das kostet Zeit. Auch die Beschichtung der Selektionsröhrchen mit dem Zielprotein ist nicht so einfach. Die Bindungschemie kann das Zielprotein verziehen oder falsch ausgerichtet an die Wand kleben, die Plastikwaren können einem weitere Probleme verursachen. Das kann durchaus dauern, bis so ein System sauber läuft und diese Probleme ausgeräumt sind. Manchmal kann man die Probleme auch einfach nicht lösen.
Gehen wir einmal davon aus, alles hat funktioniert, das Protein hat die Füßchen des Phagen so ersetzt, dass sie als neue Füßchen funktionieren. Die Phagen, die mit den neuen, mutierten Füßchen an das Ziel binden, bleiben an diesem Ziel kleben, die restlichen kippt man weg (vorzugsweise in ein verschließbares Gefäß, da anschließend autoklaviert (also hitzesterilisiert) wird. Also NICHT in den Ausguss kippen!).
Diese gebundenen Phagen, „kratzt“ mach mittels Waschlösungen (höherer pH-Wert, mehr Salz, was auch immer funktioniert, muss man halt ausprobieren), von der Wand und vermehrt sie in ihrem Wirt (meist E.Coli). Dann reinigt man die Phagen aus E.Coli (experimentelle Details erspare ich mir, das steht in der Gebrauchsanleitung des Phagenkits) schnippelt sein selektiertes Gen raus (mit den üblichen Labormethoden), sequenziert einige davon (nachdem man sie vereinzelt hat, Stichwort Phagenplaques, auch hier, dafür gibt es die Kochrezepte in Molecular Cloning[vi], das werde ich hier nicht ausführen).
Nun fängt das Spiel von vorne an.
A) Man hat in seinen selektierten Genen nur wenige, spezielle Varianten, die sich wiederholen. Man braucht mehr Diversität als noch mal ErrorProne PCR.
B) Man hat eine hohe Diversität hier fängt man an, diese verschiedenen Mutationen miteinander zu kombinieren, damit sie sich gegenseitig verbessern und unterstützen, weil, gemeinsam sind wir stark auch als Punktmutationen!
Sollte B eingetreten sein kommt man zum Schritt DNAShuffling.
=== Schritt 3: Ich mixe mir mein Gen ===
DNAshuffling simuliert salopp gesagt Sex im Reagenzglas. Das, was beim Sex bei den Chromosomen passiert, das sogenannte crossing-over[vii], welches bedeutet, dass sich die DNA Stränge in Sachen Geninformation mischen und kombinieren. Genaueres kann sich jeder auf der Webseite in der Fußnote durchlesen, das würde jetzt zu weit führen hier noch mal Schulstoff komplett zu wiederholen.
„Simuliert“ wird das im Reagenzglas eher auf die brutale Art und Weise (also mit deutlich weniger Spaß für die Beteiligten als beim natürlichen crossing-over).
1. Man hackstückelt sein Gen in handliche Fragmente von 50-200 Basenpaaren. Dafür kann man Enzym/Eiweiß namens DNAse verwenden, die DNA wahllos verhackstückelt (schwer zu kontrollieren) oder eine chemische Methode, die ich in meiner Promotion entwickelt haben namens (NExT) DNA shuffling [viii] (ja, schamlose Eigenwerbung. Ich habe die Drecksarbeit an der Bench gemacht, mein Doktorvater hat sich aber den Erstautor genommen und seiner Frau den Letztautor, also die Gruppenleiterposition gegeben. Unschöne Geschichte). Die experimentellen Details kann jeder selbst nachlesen, den es interessiert.
2. Diese kleinen Fragmente reinigt man mit den üblichen, bekannten Methoden.
3. Man setzt die Fragmente wieder zusammen, zufällig und zwar mittels Tataaaaaaaaaaaaa PCR. Wer hätte das gedacht? Diese PCR nennt sich total kreativ WiederzusammensetzPCR oder halt auf Englisch Reassembly PCR.
Und jetzt wird es spannend…
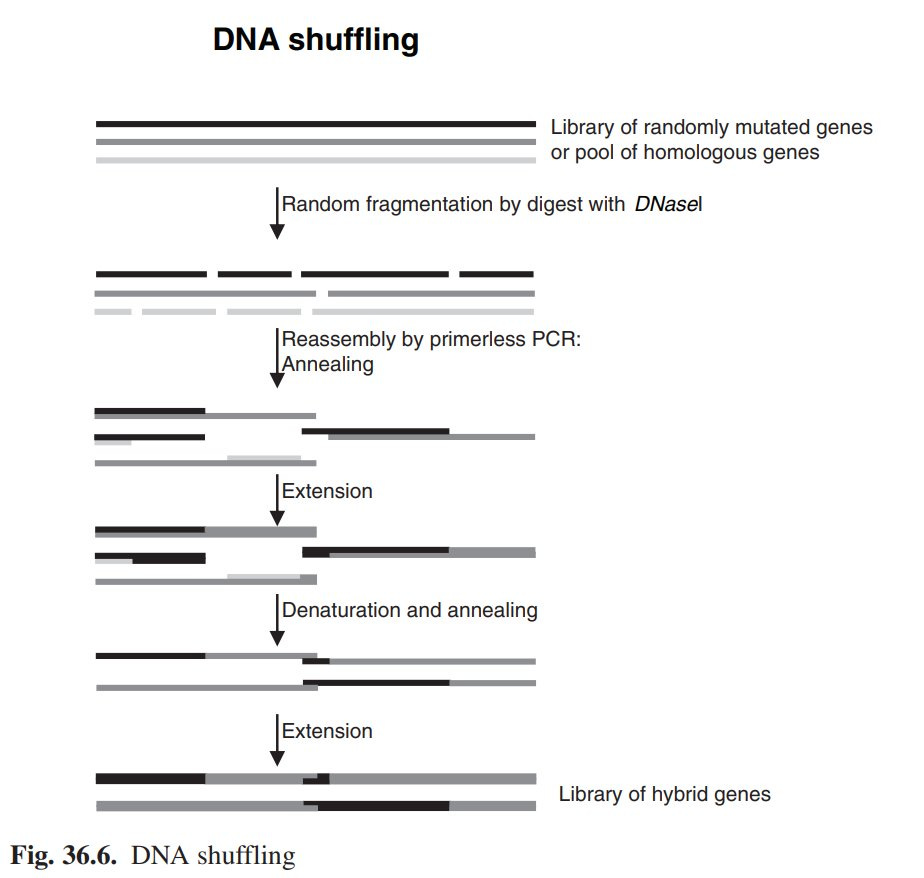
Das Schaubild stammt aus einem meiner alten Buchkapitel[1]
Die kleinen DNA Fragmente dienen sich dabei gegenseitig als Primer und verlängern sich spontan gegenseitig wieder zum ganzen Gen (mit einigen Unfällen und Fehlern, aber es kommt einiges an wiederzusammengesetztem korrektem Gen dabei heraus).
Anmerkung: An dieser Stelle kann man z. Bsp. auch zusätzliche Fragmente wie eine Furin-Schnittstelle in die Mischung werfen und schauen, ob und wo sie integriert und trotzdem noch ein funktionsfähiges Protein entsteht.
Irgendwann wird man zum Screenen der Furin Schnittstelle dann schon in Zellkultur und Pseudoviren wechseln müssen, schätze ich. Bei Zellkultur bin ich aber raus, habe ich nie gemacht.
Diese wieder zusammengesetzten Gene kopiert und vermehrt man mittels PCR (schon wieder *gähn*), weil die Menge einfach zu klein ist, um damit gescheit arbeiten zu können und packt die in seinen Phagen der Wahl. Und das Spiel beginnt von vorne.
Ist das System robust und sauber aufgesetzt, schafft man 1-2 Runden Evolution pro Woche.
====Kleiner Coronatest Exkurs====
Warum ist die Fehlerrate bei der Corona PCR möglicherweise so hoch?
1. Sie verwenden Taq mit Mangan im Puffer (geschenkt).
2. Es ist eine 2 Stufen PCR. Der abgekratzte RNA Trümmerhaufen aus den geschundenen Nasen wird erst einmal in zufällige DNA-Fragmente übersetzt und anschließen lässt man da Taq und Primer drauf los… Erkennt ihr die Parallelen zum DNA-Shuffling und zur Reassembly PCR?!
Wer mag, darf den Text gerne ins Englische übersetzen, ich bin zu faul dazu.
[1]https://www.researchgate.net/publication/227206563_Directed_Protein_Evolution
[i]https://www.researchgate.net/publication/227206563_Directed_Protein_Evolution
[ii]https://www.gesetze-im-internet.de/wisszeitvg/BJNR050610007.html
[iii] Wilson DS, Keefe AD. Random mutagenesis by PCR. Curr Protoc Mol Biol. 2001 May;Chapter 8:Unit8.3. doi: 10.1002/0471142727.mb0803s51. PMID: 18265275.
[iv] https://de.wikipedia.org/wiki/Bakteriophagen
[v] https://de.wikipedia.org/wiki/Phagen-Display
http://molecularcloning.com/
[vii] https://www.biologie-seite.de/Biologie/Crossing-over
[viii] Müller KM, Stebel SC, Knall S, Zipf G, Bernauer HS, Arndt KM. Nucleotide exchange and excision technology (NExT) DNA shuffling: a robust method for DNA fragmentation and directed evolution. Nucleic Acids Res. 2005 Aug 1;33(13):e117. doi: 10.1093/nar/gni116. PMID: 16061932; PMCID: PMC1182171.
[i] https://www.projectveritas.com/news/pfizer-executive-mutate-covid-via-directed-evolution-for-company-to-continue/