

Ziel der folgenden Zusammenfassung ist es, eine Grundlage für Presseberichterstattung anhand bekannter Daten zu liefern und aufzuzeigen, in welchem Ausmaß die Datenlage mit anderweitigen Verlautbarungen/Informationen seitens des PEI nicht in Übereinstimmung zu bringen ist.
I. Chronologie, mitgeteilte Daten
Ende 2020 – dies ist öffentlich bekannt – startet das PEI die SafeVac2.0 App. Voraussetzung für die Registrierung ist die Angabe einer gültigen Charge, d.h. ALLE Teilnehmer haben wenigstens eine Impfung.
Am 30.09.2022 – nach Verlängerung um 9 Monate – wurde die Studie für die Aufnahme neuer Teilnehmer geschlossen. Das Studienende zu diesem Datum ist der Internetseite des PEI zu entnehmen und auch der BT-Anfrage Arbeitsnummer 6/315 vom 03. Juli 2025.
Am 18.08.2023 veröffentlicht das PEI eine Stellungnahme1
Dort heißt es: Insgesamt registrierten sich 734.394 Personen für die Teilnahme an der SafeVac 2.0-Studie.

Einige Teilnehmer hatten eine 2. Dosis. Der Anteil der 2fach geimpften Teilnehmer von allen Studienteilnehmern betrug: 445.483 von 734.394, entsprechend 60,7 Prozent.
Dieser Wert wurde berechnet, weil sich hieraus indirekt auf die anzunehmende Zahl der Teilnehmer mit Comirnaty schlussfolgern lässt, siehe unten. Es wird davon ausgegangen, dass dieser Anteil weitgehend impfstoffunabhängig war.

Unter dieser Annahme errechnet sich eine Comirnaty-Teilnehmerzahl von ungefähr 437.000 bis 438.000 Teilnehmer.2
Bezogen auf die Gesamtzahl der Teilnehmer von 734.394 entspricht der anzunehmende Comirnaty-Anteil bei den Teilnehmern somit 59,7 Prozent. Der Erwartungswert wäre tendenziell aufgrund der Marktanteile etwas höher.
Für diese ca. 60 % der SafeVac2.0-Teilnehmer, die mit dem Impfstoff Comirnaty geimpft worden sind, wies das Paul-Ehrlich-Institut in der Meldung vom 18.08.2023 folgende Werte für „schwerwiegende“ Ereignisse einschließlich AESI aus:

Die Liste der AESI, bei denen es sich allerdings häufig ZUGLEICH um schwerwiegende Ereignisse handeln dürfte, sieht wie folgt aus lt. PEI hinsichtlich Meldehäufigkeiten3, dort S. 21.
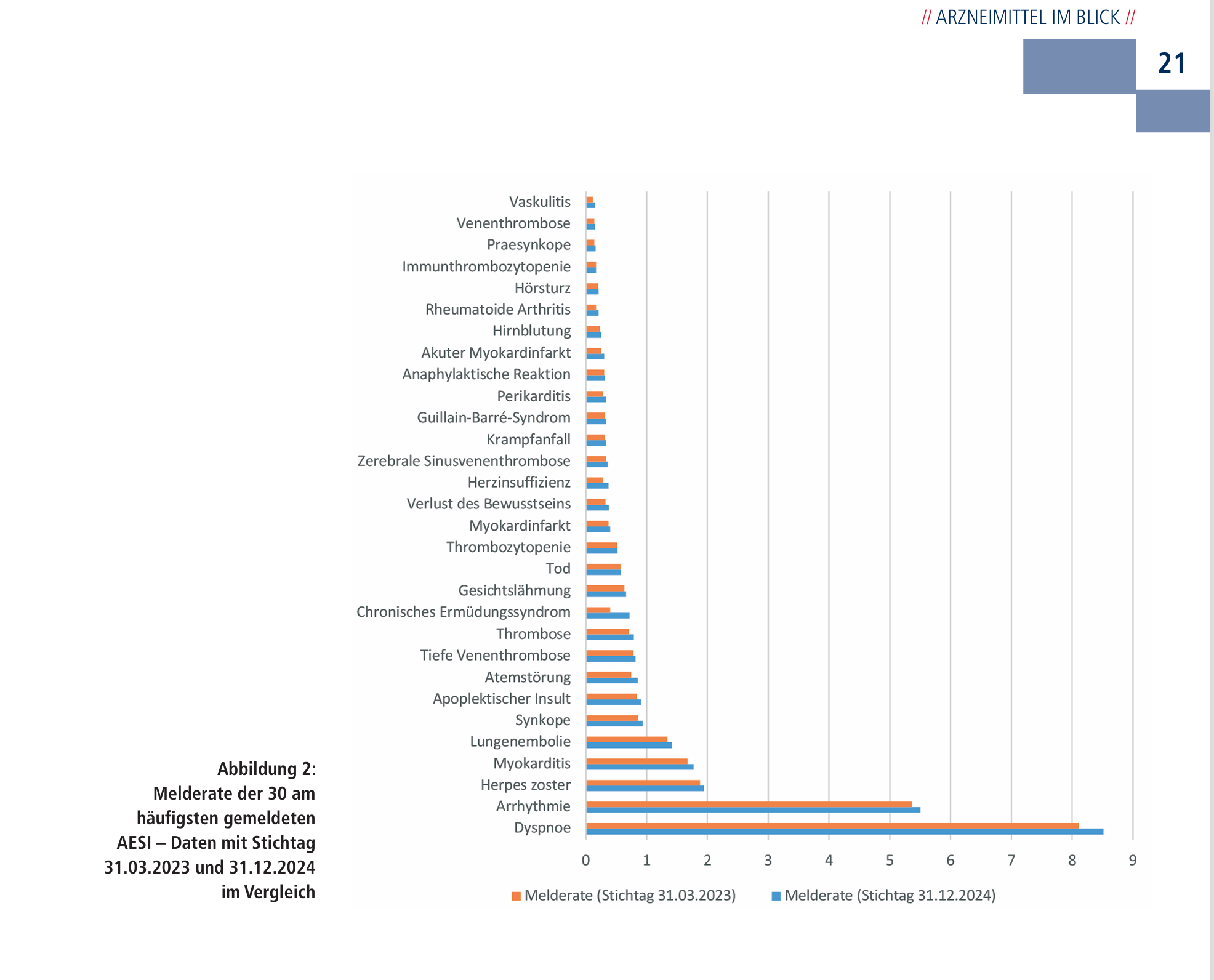
Aus obigen Daten, da Comirnaty nur ca. 60 % aller SafeVac2.0 Teilnehmer repräsentiert, würde sich ein Erwartungswert von 3.935/60*100 ergeben, mithin eine zu erwartende Anzahl schwerwiegender Ereignisse bzw. Verdachtsfälle von 6.558.
In der Bundestagsantwort vom 03.07.2025 wird entgegen der bisherigen Systematik der PEI-Mitteilungen auf die Teilnehmeranzahl zurückgegriffen, nicht auf die Anzahl der Verdachtsfallmeldungen mit schwerwiegendem Charakter. Einzelne Teilnehmer können durchaus mehrere schwerwiegende Ereignisse bei 2. oder sogar 3. Impfung erlebt haben. Die Frage bezog sich auf die Zahl der Verdachtsfälle (nach einzelnen Impfungen, denn nach weiterer Impfung ergibt sich gundsätzlich ein NEUER Verdachtsfall, wenn wiederum adverse events auftreten). Die Frage wurde somit nicht beantwortet, wobei die Frage zugleich auf ALLE Verdachtsfälle, nicht nur auf die schwerwiegenden Verdachtsfälle gerichtet war.


Unklar ist anhand der Antwort, ob die Zählung laut der Bundestags- Antwort vom 03.07.2025 die AESI mit beinhalten soll, wobei es sich (s. o.) bereits in vielen Fällen per definitionem um zugleich schwerwiegende Ereignisse handelt, z. B. im Falle von Lungenembolien, Synkopen, apoplektischem Insult, Thrombosen, Tod, Myokardinfarkt usw.
Auffallend ist jedenfalls, dass sich in Relation zur zu erwartenden Anzahl der SAE (einschl. AESI) ein auffälliges Missverhältnis ergibt. Der Erwartungswert (6.558) wird mit der Angabe 3.506 teilnehmende Personen deutlich unterschritten, wenn man unterstellt, dass die meisten Teilnehmer, die bei der 1. Impfung ein schwerwiegendes Ereignis verzeichnet haben, ohnehin kaum nochmals geimpft worden sein dürften mit einem offenbar für sie nicht verträglichen Impfstoff.
II. AMG – Meldepflicht, Prozessvortrag
§ 62 Abs. 2 AMG4 lautet:
(2) Die zuständige Bundesoberbehörde erfasst alle Verdachtsfälle von Nebenwirkungen, von denen sie Kenntnis erlangt. Meldungen von Patienten und Angehörigen der Gesundheitsberufe können in jeder Form, insbesondere auch elektronisch, erfolgen. Meldungen von Inhabern der Zulassung nach § 63c erfolgen elektronisch. Die zuständige Bundesoberbehörde stellt durch Sammeln von Informationen und erforderlichenfalls durch Nachverfolgung von Berichten über vermutete Nebenwirkungen sicher, dass alle geeigneten Maßnahmen getroffen werden, um sämtliche biologische Arzneimittel, die im Geltungsbereich dieses Gesetzes verschrieben, abgegeben oder verkauft werden und über die Verdachtsfälle von Nebenwirkungen berichtet wurden, klar zu identifizieren, wobei der Name des Arzneimittels und die Nummer der Herstellungscharge genau angegeben werden sollen.§ 62 Abs. 3 AMG5 legt die Meldefristen fest, die sich je nach Schweregrad eines Verdachtsfalles unterscheiden:
(3) Die zuständige Bundesoberbehörde hat jeden ihr gemeldeten und im Inland aufgetretenen Verdachtsfall einer schwerwiegenden Nebenwirkung innerhalb von 15 Tagen und jeden ihr gemeldeten und im Inland aufgetretenen Verdachtsfall einer nicht schwerwiegenden Nebenwirkung innerhalb von 90 Tagen elektronisch an die Datenbank nach Artikel 24 der Verordnung (EG) Nr. 726/2004 (EudraVigilance-Datenbank) zu übermitteln. Die zuständige Bundesoberbehörde arbeitet mit der Europäischen Arzneimittel-Agentur und dem Inhaber der Zulassung zusammen, um insbesondere Doppelerfassungen von Verdachtsmeldungen festzustellen. Die zuständige Bundesoberbehörde beteiligt, soweit erforderlich, auch Patienten, Angehörige der Gesundheitsberufe oder den Inhaber der Zulassung an der Nachverfolgung der erhaltenen Meldungen.Entsprechend diesen gesetzlichen Verpflichtungen hat das Paul-Ehrlich-Institut (hier aus juristischen Gründen als Beklagte = Bundesrepublik Deutschland geführt) beim Verwaltungsgericht Darmstadt vorgetragen, Az. 6 K 716/22 DA, SS vom 11.04.2023:

Es wurde mithin unter Geltung der prozessualen Wahrheitspflicht behauptet, dass die Verdachtsfälle nach § 62 Abs. 2 und 3 AMG zu EudraVigilance weitergeleitet worden und in die fortlaufende Risikobewertung „einbezogen“ worden sind, siehe Screenshot, Bl. 444 der Gerichtsakte.

Dem WIDERSPRICHT die jetzige Auskunft in der Bundestags-Antwort vom 03.07.2025:

Würde diese Auskunft zutreffen, so wären seit dem Jahresende 2020 – wobei die meisten Studienteilnehmer im Jahr 2021 und 2022 befragt worden sind – die nicht schwerwiegenden Verdachtsfälle NICHT zu EuraVigilance weitergeleitet worden. Dies würde lt. Bundesregierung (PEI) erst „mit Abschluss der Auswertung der klinischen Studie“ geschehen.
Es würde auch bedeuten, dass diese Fälle bei der Risikobewertung entgegen dem oben genannten prozessualen Vortrag NICHT in die Risikobewertung eingeflossen sind (!). Falscher Vortrag bei Gericht ist wiederum strafbar als versuchter Prozessbetrug.
III. EudraVigilance-Daten
Die EudraVigilance-Daten, hat ein Datenanalyst heruntergeladen und in auswertbarer Form ins Internet gestellt hat.6

Basierend auf EudraVigilance-Daten kann man unter o. g. Link im Menü „Country“ auswählen.
Es erscheint folgender Bildschirm:
Nach Länderauswahl für „Deutschland“ erscheint folgende Anzeige:
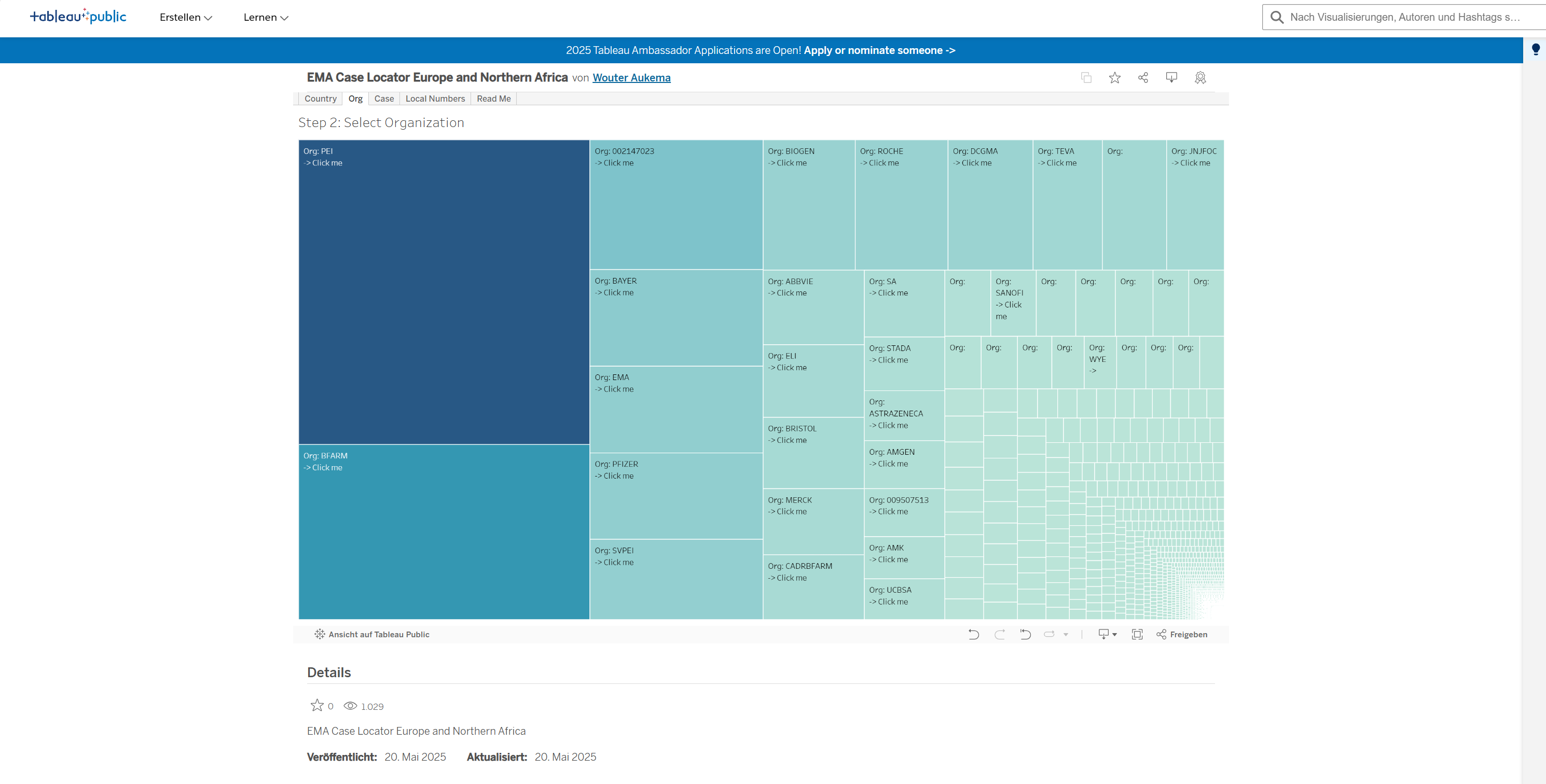
Die Größe der Kacheln symbolisiert die Anzahl der von der jeweiligen Organisation, u. a. PEI und BFarm, Merck etc. zu Eudravigilance gesendeten Meldungen. Je größer die Kachel, desto größer die Anzahl der Eudravigilance-Einträge (für alle Medikamente). Die zahlreichen kleinen Kacheln repräsentieren geringe Meldezahlen für seltener verwendete Medikamente bzw. stehen für geringe Meldeaufkommen kleinerer pharmazeutischer Unternehmen.
Unter den sendenden Organisationen befindet sich auch eine Kachel „SVPEI“.
Diese hat 56.545 Eintragungen.
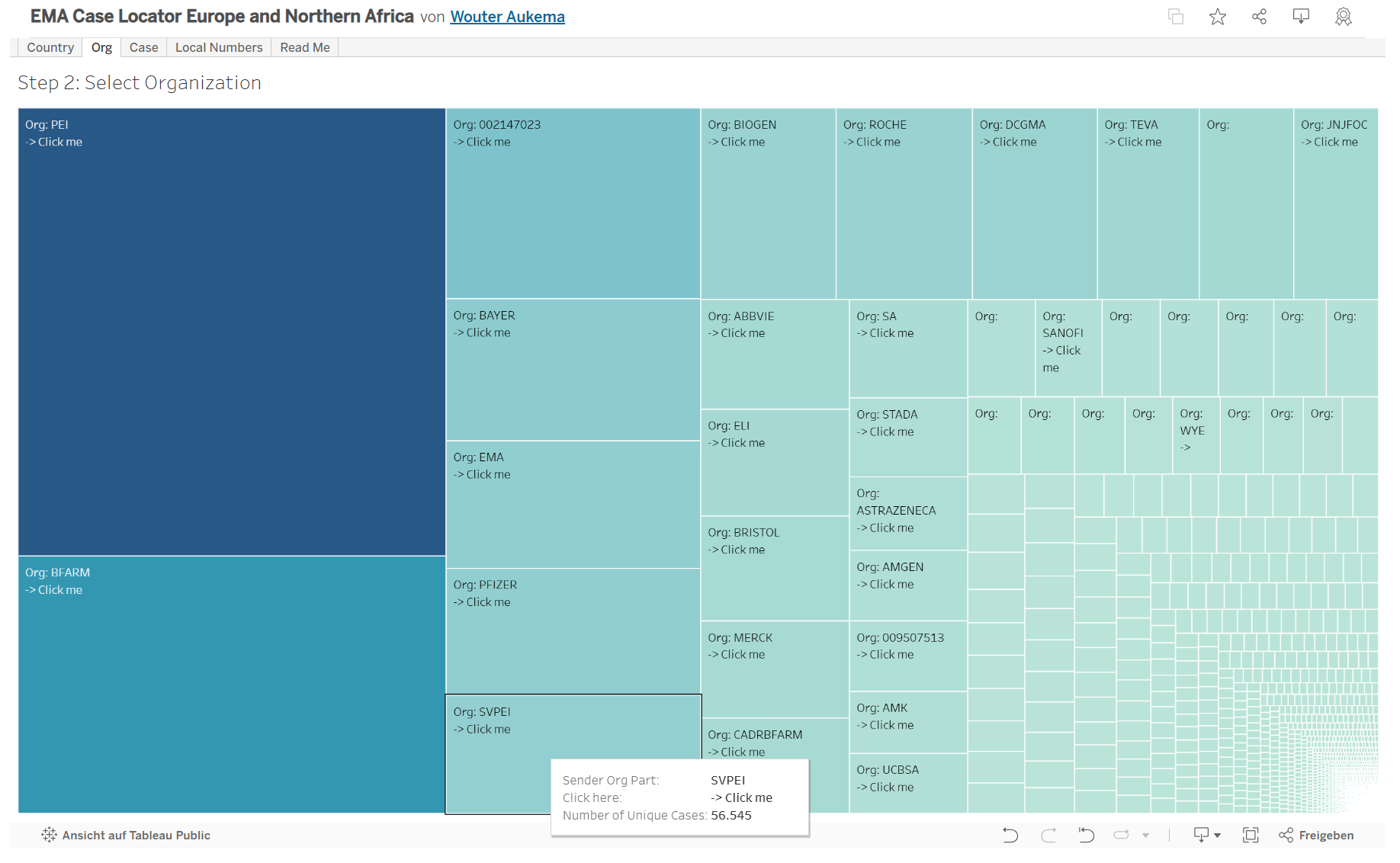
Beginnend mit dem Jahr 2020 und endend mit dem Meldejahr 2023 sind dort insgesamt 56.545 verschiedene EudraVigilance-Nummern hinterlegt:
Die Liste lässt sich als pdf generieren (590 Seiten lang), hier Screenshot aus dem pdf der letzten Seiten:
Jede Seite hat 96 Nummern, die letzte Seite eine weitere Nummer (aus Meldejahr 2023), somit 56.545 Nummern mit dem Kürzel DE-SVPEI-20xx….. (xx steht dabei für das Melde- oder Erfassungsjahr, die weiteren Zahlen sind Zuordnungsnummern).
Der Sender SVPEI steht dabei für SafeVac-PEI. Die Erfassung dieser Meldungen, wenn man eine Analyse im Detail vornimmt, erfolgt jenseits der offiziellen Statistik über Nebenwirkungen.
Auf ca. 1.18 Millionen Impfungen in SafeVac bedeutet dies, dass das PEI für ca. 4,8 Prozent der Teilnehmer PRO Impfung, eine meldepflichtige Nebenwirkung zu EudraVigilance übermittelt hat. Entsprechend hätten bei dieser NW-Rate nach der 3. Dosis 13,3 Prozent der Teilnehmer ein meldepflichtige Nebenwirkung erlebt.
Meldepflichtig ist nach § 6 Abs. 1 Nr. 3 IfSG7 der Verdacht einer über das übliche Maß hinausgehenden gesundheitlichen Schädigung:
3. der Verdacht einer über das übliche Ausmaß einer Impfreaktion hinausgehenden gesundheitlichen Schädigung,Diese Anzahl von durch C19-Impfnebenwirkungen tatsächlich betroffenen Menschen sollte offenbar auch in der PEI-Stellungnahme vom 18.08.2023 nicht erwähnt werden. Erneut wird sie jetzt in der Antwort auf die Bundestags-Anfrage vom 03.07.2025 verschwiegen.
erstellt am 03.07.2025
durch Rechtsanwältin Dr. Meyer-Hesselbarth
(Formatierung und Übertragen auf Substack von Dr. Sabine C. Stebel.
Die Bilder mussten teilweise neu erzeugt werden, da die Auflösung im Original zu schlecht für Substack war, entsprechen aber denen, die mir eingereicht wurden.)
Mathematisch genau 437.627 Teilnehmer. Dieser Wert ist eine Rechengröße, da ggf. auch Impfstoffe gewechselt worden sind etc.
§ 62 AMG - Einzelnorm https://www.gesetze-im-internet.de/amg_1976/__62.html
§ 62 AMG - Einzelnorm https://www.gesetze-im-internet.de/amg_1976/__62.html
§ 6 IfSG - Einzelnorm https://www.gesetze-im-internet.de/ifsg/__6.html
Interessant in diesem Zusammenhang was FrauHodl ausgegraben hat:
https://x.com/FrauHodl/status/1939939113240596723
"Prior to commencing clinical development, on February 6, 2020 ‼️ BioNTech obtained feedback from the Paul Ehrlich Institute ("PEI") on plans for rapid vaccine development in response to COVID-19 outbreak following a Scientific Advice Meeting."
Link auf das Dokument:
https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf
Das ergibt sich eigentlich aus den Regeln über die Zusammenführung von Verdachtsfallmeldungen. Werden weitere Beschwerden /Ereignisse/Symptome zu ein- und derselben Impfdosis gemeldet, so müssen diese im Pharmakovigilanz-System (eigentlich) durch Aktualisierung der bisherigen Meldungen ergänzt werden. Doppelmeldungen zur gleichen Medikamentengabe sind dabei zusammenzuführen. Das ganze läuft also nicht unter "neuer Verdachtsfall", sondern unter Nachmeldung und "aktualisierte Eingangsbestätigung" zu einem bestehenden Verdachtsfall, dessen Registriernummer sich nicht ändert, weil noch etwas hinzugekommen ist. Sehr anschaulich anhand von Fällen auf public tableau (Wouter Aukema) zu sehen, wo manche Fälle 12 verschiedene Eintragungen haben, aber natürlich nicht deshalb 12x gezählt werden!