


Ugurs grenzdebile Schwachsinnsideen im Protein Design
Teil 2: Warum es eine grenzdebil schwachsinnige Idee war, ein strukturell dynamisches Protein überhaupt auch nur anzufassen, dessen Struktur man bis heute noch nicht einmal im Detail kennt.

Bevor wir in das eigentliche Problem einsteigen, muss ich ein wenig Proteinfaltungsgrundwissen vermitteln, damit der geneigte Leser versteht, was das Problem in diesem Kapitel ist.
Proteinfaltung ist ein mittlerweile über 60 Jahre alter Forschungsbereich in der Biochemie. Seit über 60 Jahren versucht man also halbwegs zu verstehen, warum und wie Proteine falten und hat gerade einmal die absolut grundlegenden Prinzipen halbwegs verstanden. In der Fachliteratur schreibt man das natürlich nicht so direkt hinein, sondern man umschreibt es blumiger, euphemistischer als:
„Das Problem der Proteinfaltung fasziniert Experimentatoren und Theoretiker gleichermaßen.“ Bzw. „Das Verständnis von Proteinen auf dieser grundlegenden Ebene bietet nicht nur viele intellektuelle Anreize, sondern hilft uns auch, die Mechanismen zu verstehen, die einer normalen Proteinfunktion oder einer Fehlfunktion von Proteinen zugrunde liegen.“ [1]
Diese zwei Zitate zeigen bereits, wie „mutig“ oder vielleicht wie wahnsinnig es ist, so ein Proteinfaltungsproblem in einem vielzelligen Organismus anzugehen, der verschiedene Organsysteme besitzt, deren Proteinproduktion sich von Zelltyp zu Zelltyp auch noch unterscheiden kann.
Es gibt einige grundlegende Prinzipien, die man als Protein Ingenieur kennen sollte, wenn man sich daran wagen sollte, ein Protein zu verändern. Genau wie ein normaler Ingenieur gewisse materialwissenschaftliche Grundlagen in seinem Studium erlernt und verstanden haben sollte. Man muss die Werkstoffe, mit denen man arbeitet verstehen, um ihre Belastungsgrenzen einhalten zu können, damit das Gebäude einem nicht plötzlich und unerwartet zusammenbricht. In der Architektur gibt es dafür den Fachbereich der Statik, in anderen Ingenieurswissenschaften die jeweiligen dazugehörigen Materialwissenschaften.
Proteine sind Bausteine und somit Baumaterialien, mit denen durch Corona-Impfungen in einer ausgelagerten Produktionseinheit (Mensch/menschliche Zellen), die Spike-Proteine (das Endprodukt) hergestellt wurde. Man könnte sagen, die Pharmaindustrie hat die Produktionskosten für ihr Produkt an den Konsumenten ausgelagert. Der Konsument ist somit der Produzent und trägt in deren verquerer Logik somit möglicherweise sogar die Verantwortung für das Endprodukt.
Das Material Protein unterliegt somit einigen materiell bedingten Einschränkungen, die man kennen muss, bevor man damit arbeitet, will man nicht, dass es spektakulär schief geht.
Ich werde hier nur die Materialeigenschaften auflisten, welche für das aktuelle Problem zum Verständnis des in diesem Kapitel behandelten Problems notwendig sind. Diese Informationen sind sehr einfach und ohne große Mühe, in entsprechenden, teils sogar schon recht alten Übersichtsartikeln zu finden[2]. Es handelt sich hier um einige in der Literatur als „common sense principles“ bezeichneten Grundsätze.
Das Thema Proteinfaltung wurde bereits in Teil 1 der Reihe behandelt. Das in Kapitel 1 erworbene Grundwissen wird in diesem Kapitel somit vorausgesetzt.
1. Es gibt viele Belege, dass die teilweise Entfaltung von Proteinen über Zwischenstufen und Zwischenzustände eine wichtige Rolle bei der Regulierung biologischer Funktionen hat. Fehlfunktionen bei diesen (Ent-)Faltungsschritten führen zu Störungen im Organismus bis hin zu Krankheiten.
2. Mutationen in Proteinen neigen zur Destabilisierung sowohl der Zwischenstufen als auch des Ursprungszustandes von Proteinen. Regionen mit geringer Stabilität können somit über die Anreicherung von teilweise entfalteten Zwischenprodukten zur Fehlfaltung von Proteinen und damit zu Aggregation und Krankheit führen.
3. Proteine sind dynamische Moleküle, die Bewegungen gegenüber dem nativen Zustand ausführen müssen, um funktionieren zu können. Zwischenzustände sind das Ergebnis großräumiger Bewegungen und können daher Aufschluss die der biologischen Funktion zugrunde liegenden Mechanismen aufklären.
4. Viele Krankheiten entstehen durch Funktionsverlust von Proteinen, der durch Mutationen verursacht wird, die eine Bindungsstelle oder aktive Stelle eines Proteins stören, oder durch erhöhte Expressionswerte (Produktionsraten), die zu einer erhöhten Aktivität eines Proteins führen. Alternativ können Mutationen jedoch auch Krankheiten verursachen durch Destabilisierung der nativen Struktur oder Stabilisierung nicht-nativer Konformationen, was zu einer Anreicherung von teilweise gefalteten Zwischenprodukten führt und somit zu Aggregation der betroffenen Proteine.
5. Größere Proteine (wie das Spike-Protein), deren Faltung weniger kooperativ (störanfälliger und anspruchsvoller) ist, sind stärker gefährdet zu aggregieren oder fehlzufalten.
Diese 5 Prinzipien sagen in kürze vor allem eines:
Mutationen in einem Protein haben eine extrem hohe Wahrscheinlichkeit, dass das über jahrmillionen evolutionär optimierte Gleichgewicht zwischen verschiedenen Proteinkonformationen, sei es während der Faltung oder während seiner biologischen Aktivität, irreparabel zerstört wird und dies somit zu Krankheit führt. Die Wahrscheinlichkeit, dass eine Mutation für das Protein und somit für den Organismus von Vorteil gereicht, ist vorhanden, aber eher gering. Möglicherweise geringer als ein Sechser im Lotto mit Zusatzzahl.
Ich habe während meiner Promotion mit den 3D Strukturen(1NCO:B[3], 1Q23[4], 1PD5[5]), aus der Proteindatenbank gearbeitet, um mir irgendwelche kruden Ideen aus den Fingern zu saugen, warum mein Protein nun stabiler sein könnte. Ich habe selbst nicht an meine Hypothesen geglaubt, hatte aber nichts Besseres anzubieten und es klang eigentlich ganz intelligent. Mehr Wasserstoffbrücken, bessere hydrophobe Packung, minimale Strukturoptimierung, die üblichen Allgemeinplätze in diesem Fachbereich. Ich bin selbst überrascht, was ich damals so geschrieben habe.
Woher stammte diese 3D Proteinstruktur eigentlich, mit welcher ich mittels des swiss pdb viewers[6] bis auf Atomebene reinzoomen konnte?
Man hatte Kristalle gezüchtet. Diese Kristalle hat man dann mittels Röntgenstrukturanalyse[7] untersucht und aus dem Beugungsmuster auf die Struktur geschlossen. Ein sehr klassisches Verfahren aus den frühen 1950er Jahren, mit dem man auch die Struktur der DNA-Doppelhelix bestimmt hat. Das war die Geschichte mit dem von Watson und Crick gestohlenen Röntgenfilm von Rosalind Franklin, da gibt es sogar ein Theaterstück dazu und ein gängiger Witz in der Genetik war: „Was haben Watson und Crick entdeckt? – Das Röntgenbild von Rosalind Franklin.[8]“
Röntgenstrukturanalsye von Proteinen ist also zu meiner Zeit der gängige Goldstandard gewesen, den ich auch für das Spike Protein erwartet hätte.
Als ich mich aber in der Proteindatenbank auf die Suche machte und erst einmal routinemäßig nur Röntgenstrukturen wollte, bekam ich genau 0 Treffer. 0 Treffer!!!
Nicht einmal Ralph Baric (der mit den Feldermausviren und gain-of-funktion Experimenten in Wuhan) hatte für seine Spike Proteinstrukturen ordentliche Kristalle gezüchtet. Sogar er hat sogenannte Cryo-Elektronenmikriskopie (Cryo-EM) gemacht.
Warum hat man Cryo-EM gemacht und nicht einfach klassisch Kristalle gezüchtet? Diese Frage hätte man sich zu beginn der Planung des Impf-Spikes einfach mal vorsichtshalber stellen sollen und in einen entsprechenden Übersichtsartikel schauen sollen, der es einem erklärt[9]:
„Leider lassen sich Membranproteine bekanntermaßen nur schwer zu geordneten Kristallen züchten, da sie […] flexibel sind und keine hydrophilen (wasserliebenden) Bereiche aufweisen […].“
Flexible Proteine sind also ein Problem bei der Kristallzucht. Wenn ein großes Protein sich also nicht kristallisieren lassen will, sollte man daran denken, dass es sich möglicherweise um ein sehr flexibles Protein handeln könnte.
Das wäre der Augenblick, bei dem alle Warnlampen eingehen sollten, wenn man sich an die oben benannten grundlegenden Prinzipien erinnert.
Flexible Proteine sind ähnlich wie Maschinen mit vielen beweglichen Teilen und Zahnrädern, die optimal miteinander intergieren müssen, damit die Maschine funktioniert. Ist eines der beweglichen Teile durch Mutation gestört, kann das die Maschine zerstören, wie ein festgefressener Kolben einen Motor. Ist die Struktur der Maschine durch Mutation und die daraus resultierenden sogenannten Langstreckeneinflüsse verzogen, ist das wie ein verzogener Rahmen eines Unfallwagens unter Umständen ein langfristig ernsthaftes Problem, welches in diesem Fall zu Krankheit führen kann.
Pfizer/BioNTech verweisen in ihren Unterlagen an die FDA und EMA übrigens auf Cryo-EM Daten. Veröffentlich wurden diese referenzierten Daten aber erst im Februar 2020[10]. An dieser Stelle frage ich mich schon, mit welcher Proteinstrukturdatei wurde beim angeblich zum Einsatz gekommenen rational Design und Planung des Impf-Spike im Januar 2020 eigentlich gearbeitet, wenn es noch keine Struktur gab, mit der man arbeiten konnte?
Cryo-EM ist, aus meiner Sicht aus mehreren Gründen problematisch, um die Struktur eines Proteins zu bestimmen. In normalem wissenschaftlichen Umfeld sicherlich kein großes Problem und durchaus OK. Wenn man jedoch ein neues, körperfremdes Eiweiß in einem Menschen produziert, wäre mir Cryo-EM viel zu ungenau.
Man arbeitet mit vielen, vielen unscharfen Elektronenmikroskopischen Fotos des Proteins und rechnet diese zu einem Modell indem man diverse Algorithmen über dieses Model schickt und damit eine Struktur herbeirechnet.
Davon abgesehen, dass man nicht unbedingt alle Blickwinkel des Proteins fotografiert bekommt, weil das Protein eine bestimmte Liegeposition bevorzugt, sind die verwendeten Algorithmen natürlich mit einem potentiellen BIAS behaftet. Es kann nur ein näherungsweises Bild der Realität erzeugt werden. Das liest sich dann sehr klugscheißerisch in den Pfizer Unterlagen wie folgt:
“Das Atommodell aus PDB ID 6XR8 (Cai et al., 2020) wurde starr in die Dichtekarte eingepasst, dann flexibel mittels Phenix unter Verwendung von Realraumverfeinerung abwechselnd mit manueller Modellierung mit Coot angepasst. Die Statistiken zur Datenerfassung, 3D-Rekonstruktion und Modellverfeinerung sind in Tabelle 1 aufgeführt” (The atomic model from PDB ID 6XR8 (Cai et al, 2020) was rigid-body fitted into the map density, then flexibly fitted to the density using real-space refinement in Phenix (Adams et al, 2010) alternating with manual building in Coot (Emsley et al, 2010). Data collection, 3D reconstruction and model refinement statistics are listed in Table 1.)[11]
Im Klartext: Wir haben und die schönsten unscharfen elektronenmikroskopischen Fotos genommen, dann ein anderes Modell namens 6XR8[12] von einer anderen Forschergruppe aus der pdb Datenbank genommen, welches erst am 11.7.2020 in die Proteindatenbank hochgeladen wurde, und darüber haben wir dann abwechselnd verschiedene Modellierungsprogramme laufen lassen und ein wenig von Hand nachgebessert, bis uns das Modell in den Kram passte.
So traurig ich auch bin euch diese Nachricht überbringen zu müssen:
KEINER WEISS AKTUELL WIE DAS SPIKE-PROTEIN AUSSIEHT!
Pfizer wusste es zum Zeitpunkt der Impfstoffherstellung erst recht nicht.
Das Bild, welches wir vom Spike haben ist wie ein Dürer Stich eines Nashorns.
Zum Vergleich ein Foto eines Nashorns. Die Unterschiede sind offensichtlich:

Man erkennt bei Dürer, dass es sich um ein Nashorn handeln muss, damit endet die biologische Verwertbarkeit des Bildes aber auch.
Es gibt mittlerweile eine Publikation, welche die diversen Cryo-EM Strukturen, die erzeugt wurden, auf einen Nenner zu bringen versucht[13]. Diese Publikation enthält einige bemerkenswerte Feststellungen:
1. Es handelt sich beim Spike-Protein um ein hochflexibles Protein mit mobilen Bestandteilen. – Es ist somit klar, dass es sich um eine sehr empfindliche Maschinerie mit vielen mobilen Teilen handelt, die fein aufeinander abgestimmt agieren müssen und die höchstwahrscheinlich evolutionär sehr fein aufeinander abgestimmt sind.
2. Trotz einer allgemeinen Ähnlichkeit der Organisation der Untereinheiten wurden bei den verschiedenen in der Datenbank hinterlegten Strukturen festgestellt, dass S-Proteine unterschiedliche Konfigurationen aufweisen. – Es gibt sehr viele Cryo-EM Strukturen und die kommen alle irgendwie zu sehr unterschiedlichen Ergebnissen. Woher will BioNTech/Pfizer dann wissen, ob ihre Struktur richtig oder näher an der Wahrheit ist?
3. Obwohl diese Strukturen wesentliche Informationen zur Identifizierung der relativen Anordnung dieser Domänen geliefert haben, ist das Ausmaß, in dem die konformative Heterogenität durch Mutation während der natürlichen Evolution des Virus und im Zusammenhang mit der Entwicklung von Impfstoff-Immunogenen verändert wird, muss noch ermittelt werden. – Übersetzt heißt das: Wir haben keine Ahnung, was für Folgen Mutationen wie Prolin-Schloss oder Varianten Anpassung auf die Struktur des resultierenden Proteins haben. Das ist aber ein spannendes Forschungsthema für die Zukunft. Dumm nur, dass man das bereits im Versuch direkt im Menschen umgesetzt hat.
4. Unsere Ergebnisse offenbaren eine heterogene Konformationslandschaft des SARS-CoV-2-Spike, die sehr anfällig für Veränderungen durch die Einführung von Mutationen an den Kontaktstellen zwischen den S1 und S2-Untereinheiten sind. – Zwischen S1 und S2 liegt die Furinschnittstelle, die von Moderna patentiert ist unter Patent Nummer US 9587003 B2[14] [15].
5. Große Veränderungen in der S-Proteinstruktur können durch wenige Mutationen entstehen.- Fehlerhaftes ablesen durch das verwendete N1-Methylpseudouridin könnte ein echtes Problem sein. Das N1-Methylpseudouridinproblem wird aber separat behandelt.
Oder, um es mit einem Review zu sagen: fehlende Bereiche sind in weiß eingezeichnet:
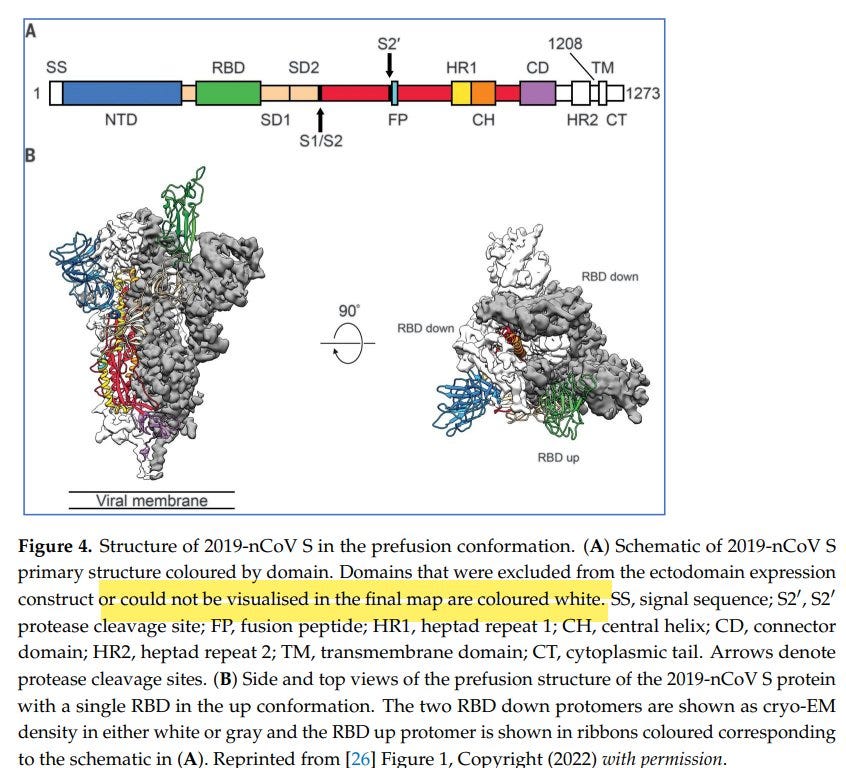
[17]
Fazit zu Teil 2:
Man hat sich an eine sehr dynamische, molekulare Maschine gewagt, deren Bauplan man im Detail nicht kennt, aber von außen grob abschätzen zu können glaubt, wie die Maschine innen aussieht.
Man hat Milliarden von Menschen ein Protein durch ihre eigenen Köperzellen bauen lassen, von dem man nicht weiß, wie es aussieht und nicht im Detail verstanden hat, wie es wirklich funktioniert. Zudem hat man dieses Protein noch durch die in Teil 1 bereits erklärte Codonoptimierung in Richtung Fehlfaltung getrieben, wofür große, flexibel-dynamische Proteine ganz besonders anfällig sind.
Einen einzigen, 10 Jahre alten, wissenschaftlichen Übersichtsartikel zu lesen, hätte vollkommen gereicht, das Problem zu erkennen Vielleicht wäre man dadurch zu der Erkenntnis gekommen, dass es eine saudumme Idee ist sich an ein großes, dynamisches, flexibles Protein heranzuwagen, dessen Struktur man nicht kennt und über dessen exakten Funktionsmechanismus aktuell noch spekuliert wird, während es von den behandelten Menschen in unbekannten Mengen hergestellt wird.
Tsytlonok M, Itzhaki LS. The how's and why's of protein folding intermediates. Arch Biochem Biophys. 2013 Mar;531(1-2):14-23. doi: 10.1016/j.abb.2012.10.006. Epub 2012 Oct 23. PMID: 23098780. https://pubmed.ncbi.nlm.nih.gov/23098780/
OK, 2 Artikel:
García-Nafría J, Tate CG. Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu Rev Pharmacol Toxicol. 2020 Jan 6;60:51-71. doi: 10.1146/annurev-pharmtox-010919-023545. Epub 2019 Jul 26. PMID: 31348870. https://pubmed.ncbi.nlm.nih.gov/31348870/
Mut? Wahnsinn? Hybris? Selbstüberschätzung? Bösartigkeit? Oder einfach nur grenzenlose Dummheit und Ignoranz?
Was sagt Ugur dazu in seinem Project Lightspeed?
"Es war keineswegs sicher, dass ein «künstliches» Spike-Protein, das im Labor losgelöst von übrigen Viruspartikeln, die es stabil halten, hergestellt wurde, jede mikroskopisch kleine Delle mit dem Spike eines Coronavirus teilen würde. Eine Abweichung um den Bruchteil einer Haaresbreite gefährdete diejenigen, die ihn erhalten. Uğur war sich dieses Risikos bewusst und suchte anhand der genetischen Sequenz und eines digitalen Modells des Virus, das er schnell erstellt hatte, nach präzisen Stellen in der Kette, an denen er das Protein «spleißen» konnte, wobei genügend der umgebenden Buchstaben – die Aminosäuren – erhalten blieben, um es zu stabilisieren, sodass es seine perfekte Form behielt (S. 48)."
Verstehe ich das richtig? Jemand, der keine Ahnung von Coronaviren hat, keine Ausbildung in Protein Engineering hat, weil er immer nur mit RNA gearbeitet hat, der grob weiß, dass das Ganze tückisch sein kann, schnippelt sich am Computer die Proteinsequenz raus und denkt "passt scho“.
Eine weitere Frage, die sich mir bei dieser Textstelle auch stellt: Ugur erstellt sich schnell ein Modell des Virus? Wie hat er das Spike-Protein anhand der Gensequenz modelliert?! Das ist laut der aktuellen Proteinfaltungsliteratur selbst mit deep learning Methoden noch nicht möglich[16].
Möglicherweise hat er ja basierend auf seinem Fernkurs Mathematik aus den 1980er Jahren ein über 60 Jahre altes Problem gelöst: Wie man anhand einer Gensequenz die 3D Struktur eines Proteins exakt und korrekt vorhersagt. Und das, ohne einen großen Rechencluster, der mehrere Tage am Stück durchrechnet. Nein, ganz alleine, daheim, am handelsüblichen Heim-PC.
Hat Ugur überhaupt eine Proteinstruktur, irgendeine Proteinstruktur aus der Proteindatenbank zu Rate gezogen?
Hatte er auch nur ansatzweise eine Ahnung, wie empfindlich die dynamischen Gleichgewichte zwischen verschiedenen Proteinkonformationen sind!?
Diese Fragen würde ich ihm gerne einmal stellen.
[1] Tsytlonok M, Itzhaki LS. The how's and why's of protein folding intermediates. Arch Biochem Biophys. 2013 Mar;531(1-2):14-23. doi: 10.1016/j.abb.2012.10.006. Epub 2012 Oct 23. PMID: 23098780. https://pubmed.ncbi.nlm.nih.gov/23098780/
[2] Tsytlonok M, Itzhaki LS. The how's and why's of protein folding intermediates. Arch Biochem Biophys. 2013 Mar;531(1-2):14-23. doi: 10.1016/j.abb.2012.10.006. Epub 2012 Oct 23. PMID: 23098780. https://pubmed.ncbi.nlm.nih.gov/23098780/
[3] Bank, R. P. D. (n.d.-a). RCSB PDB - 1NOC: Murine inducible Nitric oxide synthase Oxygenase domain (Delta 114) complexed with Type I E. coli chloramphenicol acetyl transferase and imidazole. https://www.rcsb.org/structure/1NOC
[4] Bank, R. P. D. (n.d.). RCSB PDB - 1Q23: Crystal structure of Chloramphenicol acetyltransferase I complexed with Fusidic acid at 2.18 A resolution. https://www.rcsb.org/structure/1Q23
[5] Bank, R. P. D. (n.d.-a). RCSB PDB - 1PD5: Crystal structure of E.coli chloramphenicol acetyltransferase type I at 2.5 Angstrom resolution. https://www.rcsb.org/structure/1PD5
[6] Swiss-PdBViewer: Download. (n.d.). http://www.genebee.msu.su/spdbv/text/getpc.htm
[7] Röntgenstrukturanalyse. (n.d.). Lexikon Der Chemie. https://www.spektrum.de/lexikon/chemie/roentgenstrukturanalyse/8031
[8] Wikipedia contributors. (2023d). Photo 51. Wikipedia. https://en.wikipedia.org/wiki/Photo_51
[9] García-Nafría J, Tate CG. Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu Rev Pharmacol Toxicol. 2020 Jan 6;60:51-71. doi: 10.1146/annurev-pharmtox-010919-023545. Epub 2019 Jul 26. PMID: 31348870. https://pubmed.ncbi.nlm.nih.gov/31348870/
[10] Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020 Mar 13;367(6483):1260-1263. doi: 10.1126/science.abb2507. Epub 2020 Feb 19. PMID: 32075877; PMCID: PMC7164637.
[11] https://phmpt.org/wp-content/uploads/2023/02/125742_S1_M4_4.2.1-vr-vtr-10741.pdf Seite 7
[12] Bank, R. P. D. (n.d.-d). RCSB PDB - 6XR8: Distinct conformational states of SARS-CoV-2 spike protein. https://www.rcsb.org/structure/6XR8
[13] Henderson R, Edwards RJ, Mansouri K, Janowska K, Stalls V, Gobeil SMC, Kopp M, Li D, Parks R, Hsu AL, Borgnia MJ, Haynes BF, Acharya P. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat Struct Mol Biol. 2020 Oct;27(10):925-933. doi: 10.1038/s41594-020-0479-4. Epub 2020 Jul 22. PMID: 32699321; PMCID: PMC8581954. https://pubmed.ncbi.nlm.nih.gov/32699321/
[14] Boyd, C. (2022, March 3). Study finds genetic code in Covid’s spike protein linked to Moderna patent. Mail Online. https://www.dailymail.co.uk/news/article-10542309/Fresh-lab-leak-fears-study-finds-genetic-code-Covids-spike-protein-linked-Moderna-patent.html
[15] https://patents.google.com/patent/US9587003B2/en
[16] Jisna VA, Jayaraj PB. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 2021 Aug;40(4):522-544. doi: 10.1007/s10930-021-10003-y. Epub 2021 May 28. PMID: 34050498. https://pubmed.ncbi.nlm.nih.gov/34050498/
[17] Parry, P.I.; Lefringhausen, A.; Turni, C.; Neil, C.J.; Cosford, R.; Hudson, N.J.; Gillespie, J. ‘Spikeopathy’: COVID-19 Spike Protein Is Pathogenic, from Both Virus and Vaccine mRNA. Biomedicines 2023, 11, 2287. https://doi.org/10.3390/biomedicines11082287